MICROBIOLOGY LABORATORY EXAM ONE
Study Guide for Exam One
Calculating magnification

The compound microscopes we use in our lab allow us to magnify objects up to 1000 times (with a 100X objective). We can calculate the magnification we are observing with the microscope by multiplying the magnification power of the objective, by the magnification power of the eyepieces. The magnification of the eyepieces is always 10X. Therefore, If you are using the 40X objective, the object you are viewing under the microscope would be magnified 400 times more than with the naked eye. If we do the math, we see that 10X times 40X equals 400X. The magnification of the objective is marked on the objective itself. |
|
Why is it important to follow the directions for proper microscope use in order?
Skipping or overlooking any steps could result in very costly damage to the objectives, eyepieces or other microscopic parts.

- 1) Place microscope gently on the counter at your workstation.
- 2) Ensure that the scope is away from any edges so that it will not fall or get knocked over.
- 3) Plug in the microscope.
- 4) Locate the ON/OFF switch and turn the microscope ON.
- 5) Locate the LIGHT LEVEL adjustment and turn the light on to a moderate level of brightness so you do not hurt your eyes.
- 6) Locate the 4X objective and make sure that that objective is pointing directly down toward the stage. (Failure to do steps 6, and 7 can result in the objective hitting the stage, causing breakage!) The 4X objective will have "4X" written on it. It is the shortest objective and has the lowest magnifying power of the available objectives. The 4X objective is often referred to as THE SCANNING OBJECTIVE. This is because the 4X objective ALWAYS the objective you should start with. The reason for this, is because it has a wider field of view and finding the area of interest on the slide in more manageable.
- 7) Use the COARSE FOCUS knob to move the stage all the way toward the bottom (base) of the scope (if it is not already there).
- 9) Once you have prepared your slide, you will place it on the STAGE, securing it under the two metal clips.
- 10) Locate the STAGE ADJUSTMENT knob and notice that this allows you to move the stage right and left and forward and back. You should try to center the specimen on your slide over the light source with the naked eye using the STAGE ADJUSTMENT knob.
- 11) Now with the stage all the way at the bottom and the 4X objective pointed at the slide, use both eyes to view the slide. You will almost always not see anything but a blur at this point.
- 12) While you continue viewing the slide through the eyepieces, use the COARSE FOCUS knob to slowly move the stage upward (toward the objective) until the slide comes into view.
- 13) Once the object on the slide is visible, you may then use the FINE FOCUS knob to focus the object further.
- 14) You MUST always start with the 4X objective when viewing a new slide. This is to prevent hitting the objective with the stage, and since the field of view is larger at 4X, finding your specimen or object will be much easier. Even seasoned scientists would have a very hard time finding a small object under 40X if they did not start st the 4X first.
- INCREASING MAGNIFICATION
- 15) Once you have your object fully focused at 4X, make sure the object is directly in the center of our point of view. From this point YOU WILL NO LONGER BE USING THE COARSE FOCUS KNOB!
- 16) Now you can rotate the NOSEPEICE carfeully to ensure that you are going directly to only the NEXT HIGHEST magnification only (this would be the 10X). Skipping over a magnification objective may cause the objective to hit the stage, causing damage!
- 17) If you have followed the instructions closely, your specimen should be relatively in focus and in your new (smaller) field of view. You may now use the STAGE ADJUSTMENT knob and the FINE FOCUS (NOT THE COARSE FOCUS) to center and focus the object at this new magnification. If you "lose your specimen" in the field of view, you must go down to the 4X objective and start over. "Fishing" for the object with the higher objectives can cause the objective the hit the stage, not to mention that it will be much easier to find the object with the larger field of view of the 4X objective.
- 18) In order to go to 40X, you will repeat steps 16 and 17 accordingly.
- NOTES
- --- Microscopes have a special property, called "being PARFOCAL" which means that once the object is focus and centered using the 4X, when you switch to the next highest objective, your specimen will be relatively centered and in focus. You should only have to make MINOR adjustments to the FINE FOCUS and the STAGE POSITION, in order to view your object at the higher objective. If you have difficulty, you must go back to 4X and recenter and refocus!
PUTTING AWAY MICROSCOPES
- 1) Turn off POWER switch.
- 2) Make sure the stage is all the way toward the bottom (toward the base).
- 3) Make sure that the 4X objective is pointing down toward the stage.
- 4) Wrap up and Secure Cord.
- 5) Carry with 2 hands; one on the base and the other on the body.
- 6) Be aware of your surrounding and carefully place the scope in the proper place.
- 7) Make sure you area is left clean.
|
|
|
cell culture
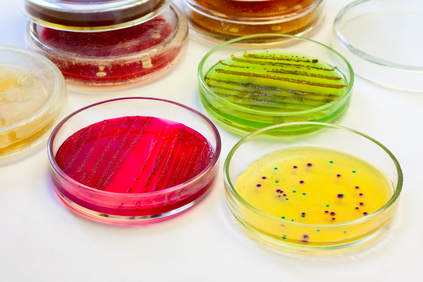
What is a "cell culture"? The definition of a "cell culture" is the process of growing cells (or cell colonies) under controlled conditions for scientific purposes. This is almost always performed in a lab, or in some other sort of environment, other than the natural environment of the microorganism.
When we study cells, it is convenient to have a lot of them to study. We can add just a few selected cells of interest into a medium with all of the yummy nutrients they need to survive and reproduce. Since microorganisms reproduce using binary fission, we end up with a lot of identical cells.
When we study cells, it is convenient to have a lot of them to study. We can add just a few selected cells of interest into a medium with all of the yummy nutrients they need to survive and reproduce. Since microorganisms reproduce using binary fission, we end up with a lot of identical cells.
Growth
Microbiologists use the term growth to indicate an increase in a population of microbes rather than an increase in size. One of the reasons prokaryotic organisms, like bacteria, have been so successful for billions of years, is that they reproduce very quickly using a process called binary fission. Binary fission creates two genetically identical daughter cells from one single parent cell. This process is more simple than mitosis, which occurs in eukaryotic cells, and it requires less energy.
Growth, in terms of microbiology, refers to the number of individual organisms that make up a population of microbes. For example, when bacteria cells are allowed to grow on an agar plate, one single bacterial cell will reproduce via binary fission and give rise to a colony or genetically identical bacteria cells that were all generated from a single parent cell.
Growth, in terms of microbiology, refers to the number of individual organisms that make up a population of microbes. For example, when bacteria cells are allowed to grow on an agar plate, one single bacterial cell will reproduce via binary fission and give rise to a colony or genetically identical bacteria cells that were all generated from a single parent cell.
|
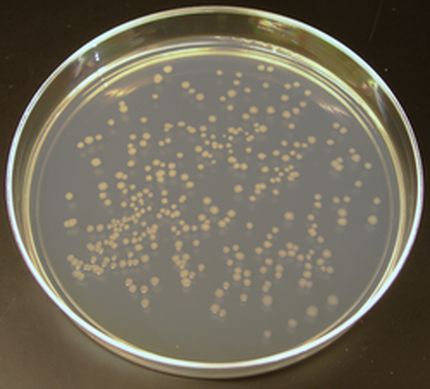
Culturing Microorganisms
The photo is of individual colonies of E. Coli cells grown on an agar plate. Microbiologists collect a small sample of microbes from a given source (we call this small sample an "inoculum") and place them into a mediumthat contains the nutrients needed for growth. This process is called "inoculation". This creates a cell culture. The medium can be in a liquid form (called broth) or in a somewhat solid, gelatin-like form (called agar/agarose). |
|

|
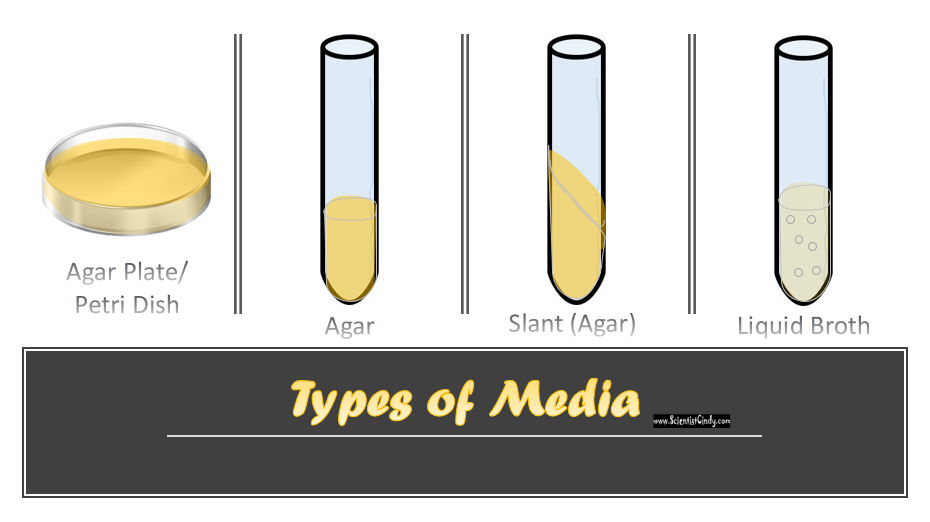
Growth Media
|

AGAR FACTS:
- Agar is a polymer made up of subunits of the sugar galactose (milk sugar).
- Agar is also a component of the cell walls of several species of red algae.
- Agar can be used in the culinary arts to thicken soups and sauces.
- Agar forms a firm gel at room temperature.
- Agar melts at approximately 85°C
- The difference in temperature from agar's melting point to the temperature at which it solidifies, 32-40°C. This is very large and make agar easy to use in the lab for preparing multiple culture plates or tubes at a time.
|
|


The most popular growth media is a nutrient-rich liquid known as LB Broth (Lysogeny broth). Some cells are able to grown free-floating in this type of media. However, most cells require some sort of surface to adhere to. For these cell, LB broth is mixed with a substance called "agar". The agar does not provide any nutrition, but it form a surface for cellular growth. Agar is added to the broth and heated in an autoclave for sterilization. When the mixture is cool enough to handle, yet hot enough to remain in liquid form, the agar can be poured into Petri dishes, culture plates, culture tubes, or test tubes. As the agar comes to room temperature, it will solidify into a firm solid/gelatinous material that has the nutrients from the LB broth.
Liquid Growth Media
Microbial growth using an agar plate will yield individual colonies of cells that have been derived from a single parent cell (ideally). This method is used in clinical laboratories to test samples of feces, saliva, cerebrospinal fluid, or blood for the presence of microorganisms.
Microbial growth using an agar plate will yield individual colonies of cells that have been derived from a single parent cell (ideally). This method is used in clinical laboratories to test samples of feces, saliva, cerebrospinal fluid, or blood for the presence of microorganisms.
Requirements for growth vary depending on the type of cell. There are a lot of different types of media available for this purpose. In general, a growth media should be in a liquid (broth) form or a semi-solid (agar plate) form.
Growth media should contain the following essential nutrients.
- amino acids
- carbohydrates
- vitamins
- minerals
- growth factors
- hormones
- CO2 or O2
- pH buffer to maintain relatively neutral pH
- osmotic pressure
- temperature (typically 37 degrees C)
|
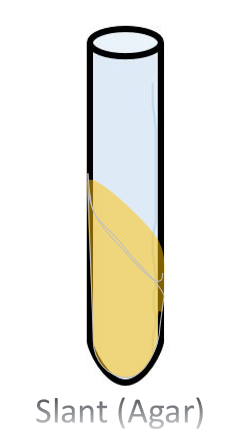
The Agar Slant Nutrient Broth and Slant Culture The agar slant, is simply a test tube in which the warm liquefied agar was poured, then the test tube was oriented in a slanted position (approximatetely 45 degree angle) and the agar was allow to cool and solidify.
|
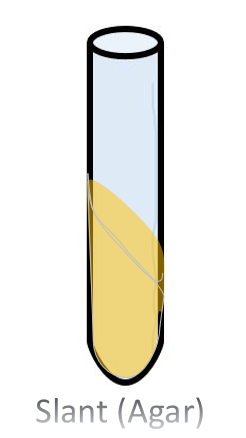
|
Aseptic Techniques
What does "aseptic" mean? The word "aseptic" means "free from microorganisms".
STEPS OF ASEPTIC TECHNIQUE
- Flame the loop or needle after each inoculation and before a new inoculation is done. This kills any leftover organism that is still on the instrument.
- An inoculation needle is used for retrieving solid or dense media. - Needles are used to transfer from solid media to other solid media or petri plates.
- An inoculation loop is used to retrieve liquid media. - • Loops are used to transfer from liquid media to liquid media or petri plates.
- TSB – Tryptic soy broth - a nutrient broth suitable for the growth of a wide variety microorganisms.
- TSA – Tryptic Soy Agar - a nutrient agar suitable for the growth of a wide variety microorganisms.
- Agar: is a natural gelatin from red algae, low melting point melting point = 100°C, solidifies around 45°C, does not contain nutrients by itself
Smear Preparations
Smear preparations are done to prepare the organism for certain types of staining techniques such as the Gram stain. Smear preparations are used to determine cell shape, arrangement of the cells.
When we do a smear prep, we fix the bacteria onto the slide.
What is the purpose of FIXATION?
Fixing the bacteria will
1) stop the decay/putrification process
2) preserve the morphology of the cells long-term
3) ensures that the cells stick to the slide, so that the cells do not fall off the slide during the staining procedure.
*In heat fixing, what would happen if too much heat were applied?
It would damage the cell's structure and can even crack the slide.
A smear preparation can be done using cells suspended in either a semi-solid (gelatinous) medium like agar or a liquid medium, such as broth.
Smear preparations are done to prepare the organism for certain types of staining techniques such as the Gram stain. Smear preparations are used to determine cell shape, arrangement of the cells.
When we do a smear prep, we fix the bacteria onto the slide.
What is the purpose of FIXATION?
Fixing the bacteria will
1) stop the decay/putrification process
2) preserve the morphology of the cells long-term
3) ensures that the cells stick to the slide, so that the cells do not fall off the slide during the staining procedure.
*In heat fixing, what would happen if too much heat were applied?
It would damage the cell's structure and can even crack the slide.
A smear preparation can be done using cells suspended in either a semi-solid (gelatinous) medium like agar or a liquid medium, such as broth.
The key to a successful smear preparation is to make sure that the smear is not too thick otherwise there may be too many cells on the slide. If there are too many cells on the slide, they pile on top of eachother and you cannot distinguish the shape or arrangement of the cells. Remember less is better.
Types of Stains
- Simple Stains - True to its name, the simple stain is a very simple staining procedure involving only one stain. An example of a "simple stain" is methylene blue.
- Differential Stain - A differential staining technique is one that uses 2 different stains.
- For example, the gram stain is considered a differential stain, because it uses more than one stain; a primary stain, and a counterstain..
- Negative Stains - The background is stained but the cells do not stain. Cells appear clear.
- Acidic and Basic Stains - If the chromophore of the dye used in the stain is positively charged, it is a basic stain.
- If the chromophore of the dye used in the stain is negatively charged, it is an acidic stain.
Some dyes are either acidic or basic.
How do you determine whether a stain is basic or acidic?
- If the chromophore of the dye used in the stain is positively charged, it is a basic stain.
- If the chromophore of the dye used in the stain is negatively charged, it is an acidic stain.
The colored ion of the dye is called the chromophore. If the chromophore is the positively charged ion, the stain is classified as a basic dye; if the negative ion is the chromophore, the stain is considered an acidic dye.

The Negative Stain
What is the Negative Stain Used For? Negative staining allows the morphology, arrangement and size of bacteria, to be viewed microscopically. How Do Negative Stains Work? Negative staining allows the morphology, arrangement and size of bacteria, to be viewed microscopically.
The cell wall of the bacteria is naturally negatively charged. A rule of chemistry, is that opposite charges attract, and "like charges" (charges that are alike) repel one another.
The result is that the bacteria cell remains unstained an appears as a clear object on the slide, while the area surrounding the bacteria cell is darkly stained by the nigrosin stain.
- Negative staining does not require fixation because the cells themselves are not being stained. Fixation can change the morphology of the cells, so the negative staining procedure preserves the morphology better.
The cell wall of the bacteria is naturally negatively charged. A rule of chemistry, is that opposite charges attract, and "like charges" (charges that are alike) repel one another.
The result is that the bacteria cell remains unstained an appears as a clear object on the slide, while the area surrounding the bacteria cell is darkly stained by the nigrosin stain.
Gram Stain
- Differential Stain - A differential staining technique is one that uses 2 different stains.
The primary stain in Gram stain is crystal violet, and the counterstain is safranin.
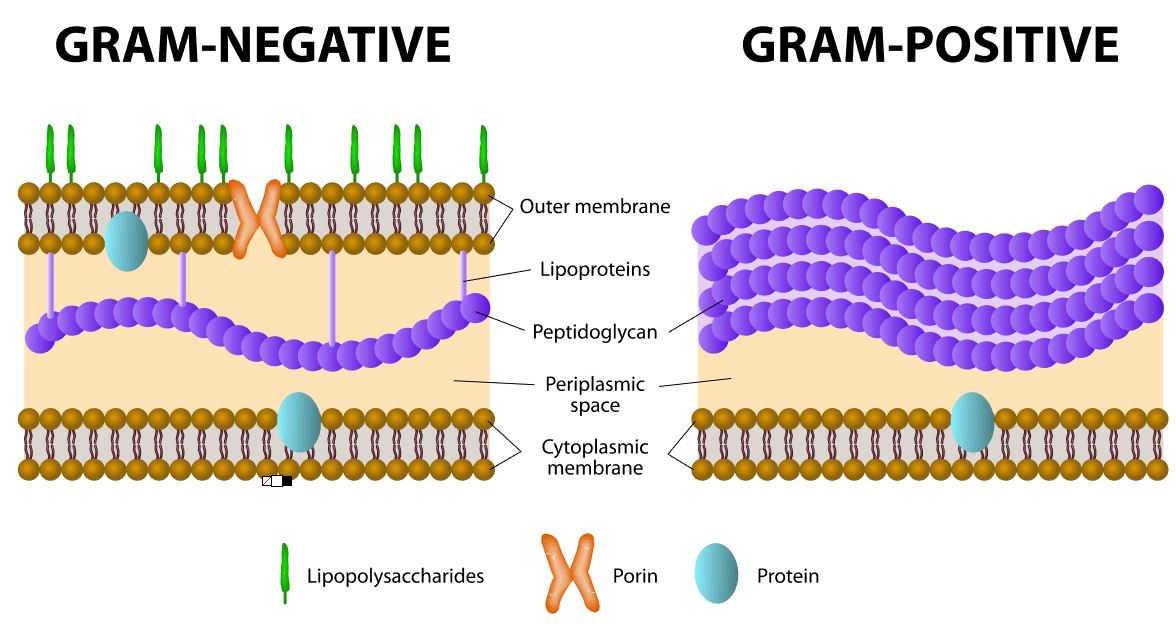
Gram stain is used to distinguish between bacteria that are gram-positive and gram-negative, due to the different cell wall structures and compositions.
Why is the Gram Stain such a BIG DEAL?
All bacteria cells that have cell walls, have peptidoglycan (either a thick layer or a thin layer).
Unlike bacteria, archaea cell walls do not contain peptidoglycan.
Many types of eukaryotic cells also have cell walls, but none of them are made of peptidoglycan
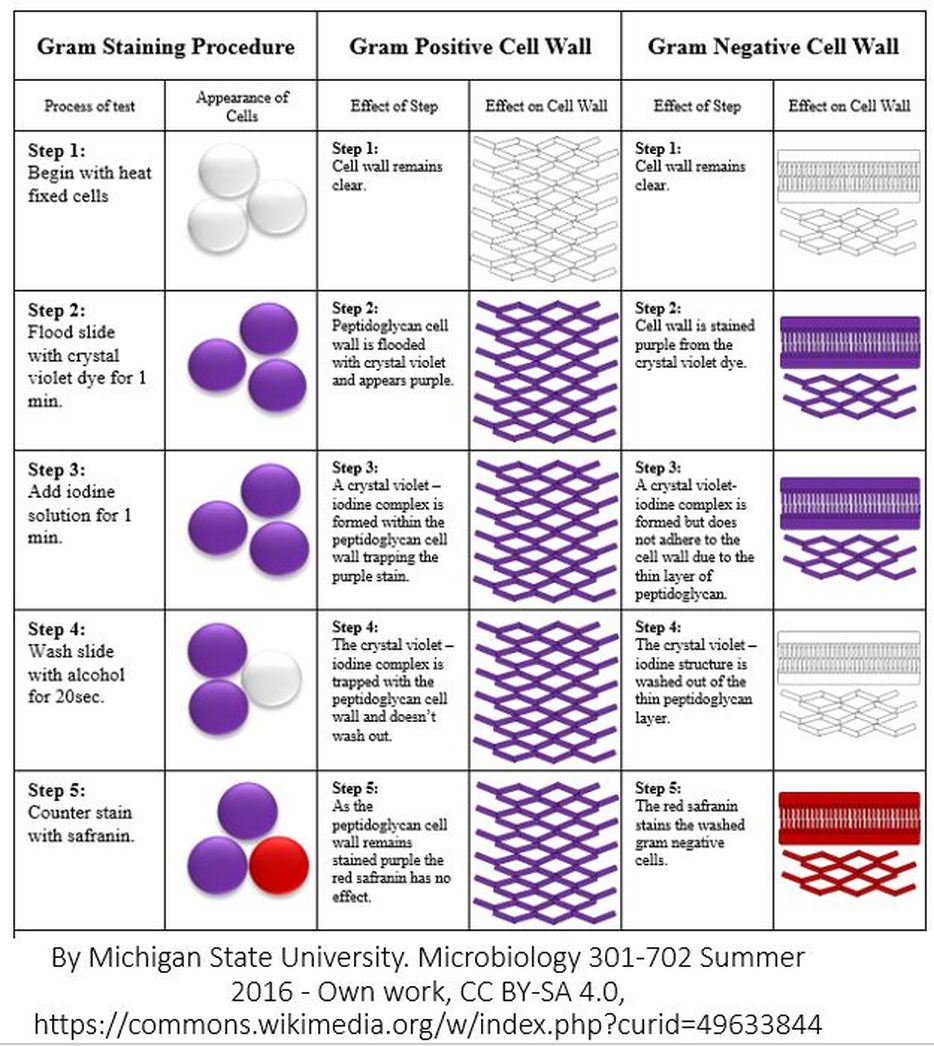
- STEP ONE = THE FIXATION STEP. The FIXATION STEP is really the last step of the smear prep from ex. 4. There are many different fixation methods. For a smear preparation, heat fixation is used, which involves briefly passing the slide over a flame. Regardless of the fixation method used, the purpose of fixation is as follows:
- to kill the bacteria
- to preserve the morphology of the cells long-term
- to help the cells adhere to the slide so that they won't wash off during the staining procedure.
- STEP TWO = PRIMARY STAIN STEP. In this step, the primary stain, Crystal Violet, is applied to the cells. Crystal Violet is a dye that colorizes peptidoglycan, which is a component of the cell wall in bacteria cells.
- STEP THREE = MORDANT STEP. A "mordant" is a substance used in staining or dying procedures that "sets" or "binds" the stain to the structure being stained. When a mordant is used, the stain is more resistant to being washed off. In the Gram stain, Iodine is used as the mordant to help Crystal Violet bind more strongly to peptidoglycan.
- STEP FOUR = THE DECOLORIZATION STEP. In the Gram stain, Alcohol is used to "decolorize" the cells. In this step some of the Crystal Violet is removed from the peptidoglycan layer. This step is 20 seconds long and it is the most important step in the Gram stain procedure. It is critical to leave the slides exposed to the decolorizer (alcohol) long enough to remove the crystal violet stain from the thin peptidoglycan layer of the gram-negative bacteria cells, but not so long that it removes all of the crystal violet from the thick peptidoglycan layer of the gram-positive bacteria cells. Exposure to the decolorizing alcohol for either too little time or too much time, will give you false results! Gram-negative bacteria have a thin peptidoglycan layer in their cell walls and should loose their purple-ish color from the Crystal Violet primary stain during this decolorization step. Gram-positive bacteria have a thick peptidoglycan layer in their cell walls and should be able to retain their purple color from the Crystal Violet primary stain following decolorization, which will during this decolorization step.
- STEP FIVE = THE COUNTERSTAINING STEP. Different counterstains can be used in the Gram stain. One of the more popular counterstains to use is called, Safranin. Safranin stains the cell wall of all bacteria cells pinkish-red. However, for the Gram-positive bacterial cells, the bluish-purple stain from the Crystal Violet overpowers the color of the Safranin and also prevents much of the Safranin from taking hold in the first place
What is the most important step of the gram procedure? The decolorizer step is the most important part of the gram stain procedure!
Why is the decolorization step the most important step of the gram procedure? Because if it is done for too long, a gram positive cell can LOOK like a gram negative cell!
If you do not decolorize long enough, then a gram negative cell can LOOK like gram positive cell.
The idea is to keep the slide in the decolorizer long enough to remove the crystal violet stain from the thin peptidoglycan layer of the gram-negative bacteria cells, but not so long that it removes all of the crystal violet from the thick peptidoglycan layer of the gram-positive bacteria cells.
Bacteria that do not have a cell wall would be considered "atypical" and will not stain at all with either Crystal Violet or Safranin!
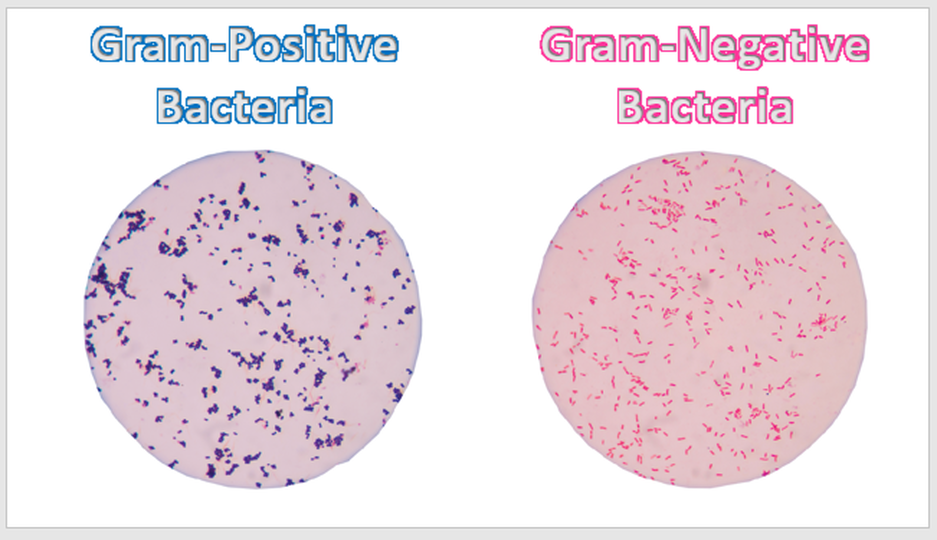
|

ATYPICAL BACTERIA
|
Sporulation
Some Gram-positive bacteria, are able to survive unfavorable environmental conditions for extended periods of time (can be hundreds of years) by forming protective endospores through a process called sporulation.
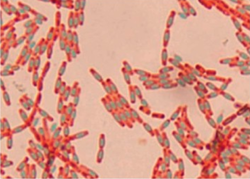
Bacteria that have endospores are existing in a semi-dormant state of existence. When environmental conditions are unfavorable, these bacteria are able to undergo sporulation. This process produces a protective endospore which allows them to go into a semi-dormant state of existence for possibly millions of years! When environmental conditions improve, the endospore is released and the cell will undergo the germination process and can return to its normal (vegetative) state.
Endospores protect the bacterial DNA from dehydration, pH, heat and radiation.
2 examples of Gram-positive bacteria that have the ability to undergo sporulation are:
Endospores protect the bacterial DNA from dehydration, pH, heat and radiation.
2 examples of Gram-positive bacteria that have the ability to undergo sporulation are:
- Bacillus
- Clostridia
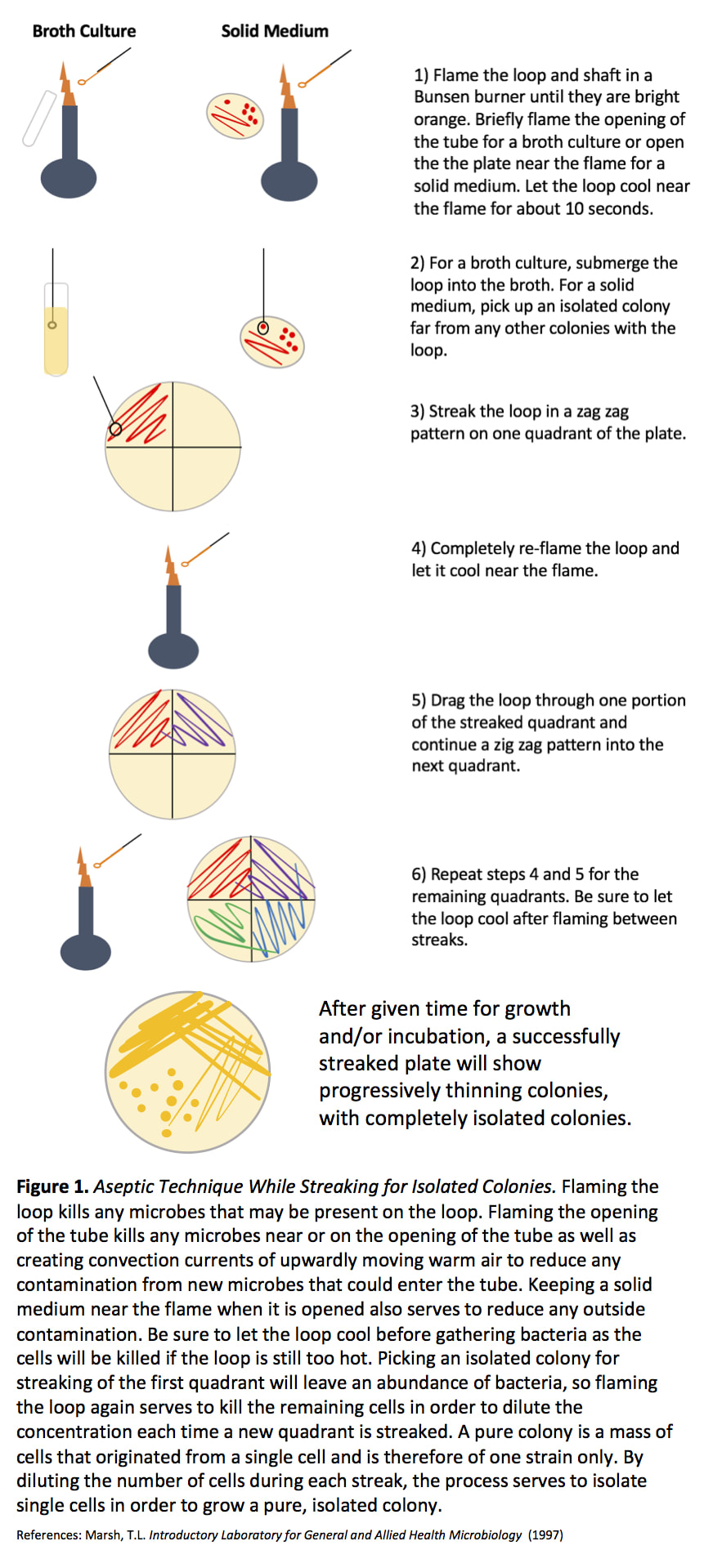
STREAK PLATE METHOD
The reason we had to heat our cells during the staining procedure, was because Endospores are difficult to stain. The heat helps the stain to penetrate the endospore.
This heating process acts as the mordant.
The primary stain in this procedure in malechite green and cells that have an endospore will appear as a pink cell (pink is due to the safranin counterstain) with a green dot inside. Why don't we see every single cell on the slide with a greem dot? It is likely that not all of the cell on the slide will have an endospore.
The endospore stain is a differential stain.
Spores are resistant to heat, dessication (drying out or dehydration), chemicals, and radiation.
In order to destroy these resilient endospores, they must be autoclaved (moist, pressurized heat) for 15 to 20 minutes at a temperature of 121°C.
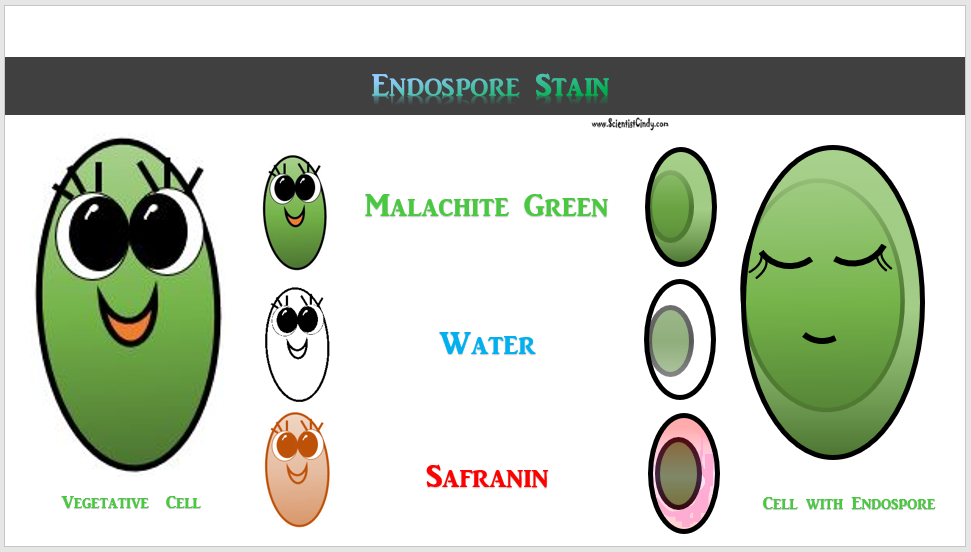
DETAILS OF THE STAINING PROCEDURE
Malachite Green - Primary Stain - The primary dye for endospore staining is malachite green.
HEAT - Mordant - Endospores are resistant to staining so HEAT acts as the mordant
Water = Decolorizer - Water acts as the decolorizer.
Safranin = counterstain - A counterstain to differentiate the vegetative cells that do not currently have an endospore
This heating process acts as the mordant.
The primary stain in this procedure in malechite green and cells that have an endospore will appear as a pink cell (pink is due to the safranin counterstain) with a green dot inside. Why don't we see every single cell on the slide with a greem dot? It is likely that not all of the cell on the slide will have an endospore.
The endospore stain is a differential stain.
Spores are resistant to heat, dessication (drying out or dehydration), chemicals, and radiation.
In order to destroy these resilient endospores, they must be autoclaved (moist, pressurized heat) for 15 to 20 minutes at a temperature of 121°C.
DETAILS OF THE STAINING PROCEDURE
Malachite Green - Primary Stain - The primary dye for endospore staining is malachite green.
HEAT - Mordant - Endospores are resistant to staining so HEAT acts as the mordant
Water = Decolorizer - Water acts as the decolorizer.
Safranin = counterstain - A counterstain to differentiate the vegetative cells that do not currently have an endospore
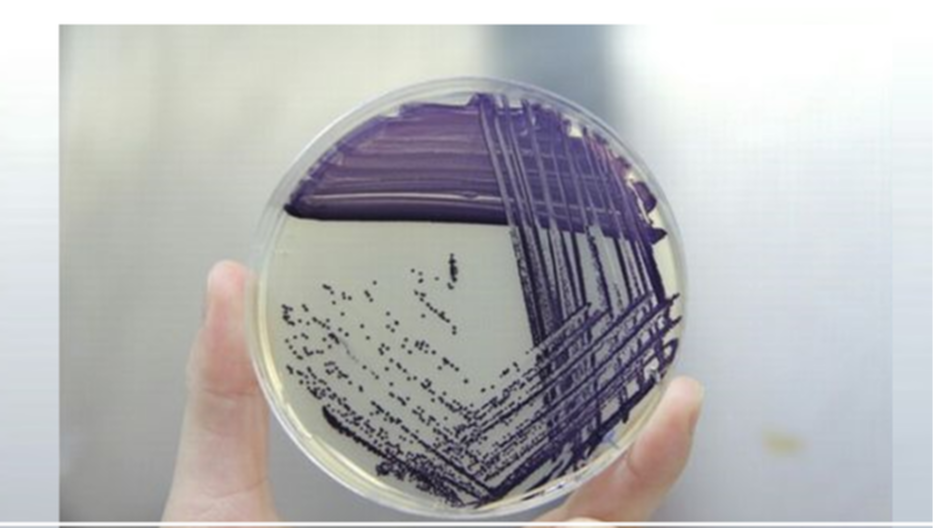
This is an example of the STREAK PLATE METHOD
Motility
If you want to view a living organism on a slide, you cannot fix it, because that would kill it. You would NOT do a smear prep, because that involves fixation.
Instead, you would do a WET MOUNT.
MOTILITY STAB TEST
For this test, a softer version of agar, called motility agar, will be used. Motility agar has a colorless dye called TTC that changes color when it comes into contact with certain types of bacteria. Some bacteria have the ability to reduce. TTC. TTC becomes red when it is reduced by these bacteria.
Instead, you would do a WET MOUNT.
MOTILITY STAB TEST
For this test, a softer version of agar, called motility agar, will be used. Motility agar has a colorless dye called TTC that changes color when it comes into contact with certain types of bacteria. Some bacteria have the ability to reduce. TTC. TTC becomes red when it is reduced by these bacteria.
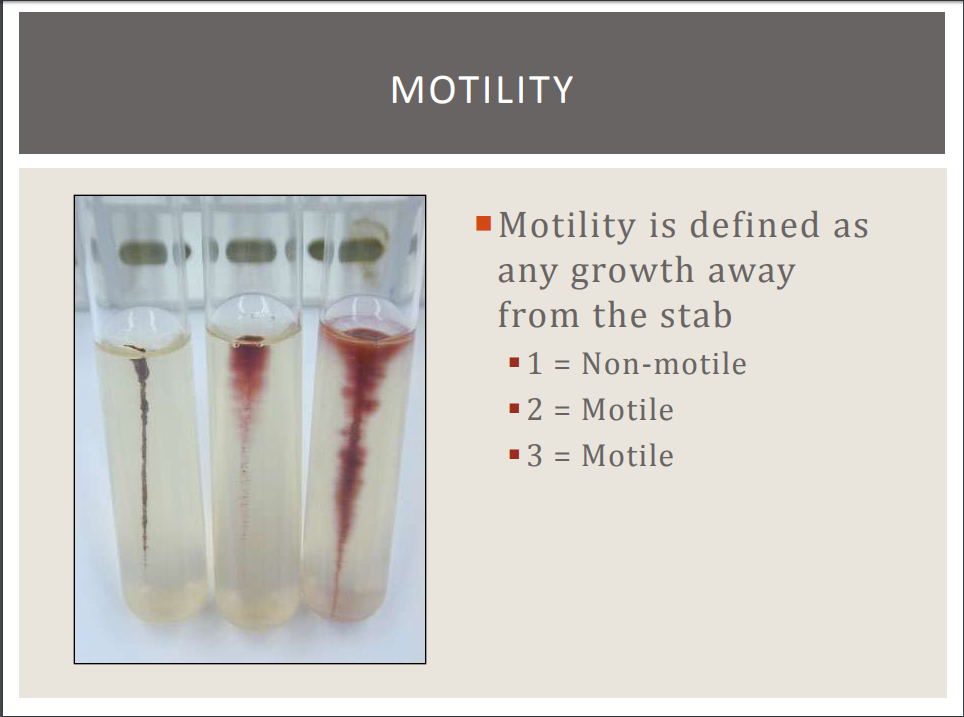
- If the bacteria is motile , and is able to reduce TTC, you can visualize the motility by noticing a red area radiating outward from a red stab-point.
- If the bacteria is NOT motile , and is able to reduce TTC, you can visualize the motility by noticing a red area radiating outward from a red stab-point.
- If the bacteria is motile , but is NOT able to reduce TTC, you can still visualize the motility by noticing a "blurry" or "fuzzy" area radiating outward from a colorlessstab-point.
- In contrast, if the bacteria is NOT motile and is NOT able to reduce TTC, then the motility agar will appear exactly as it did immediately after the stab (inoculation); as a colorless stab-point with no visible distortions radiating outward from it.
You can watch the video below for more help on concentration calculations, enumerations and dilution series!



|
A solution has 2 components.
|

|
Concentration is a RATIO that expresses the amount of solids you have to the amount of liquid you have.
So OUR Value of Concentration in Microbiology is given as…

Practice Problem #1
|
|
I have a used my entire volume of cell culture (5 ml) to create a pour plate that grew 25 colonies overnight. What was the concentration in my cell culture?
Volume = 5ml
|
Equation: Concentration = 25 cells = 5 cells/ml
5 ml

Practice Problem #2
|
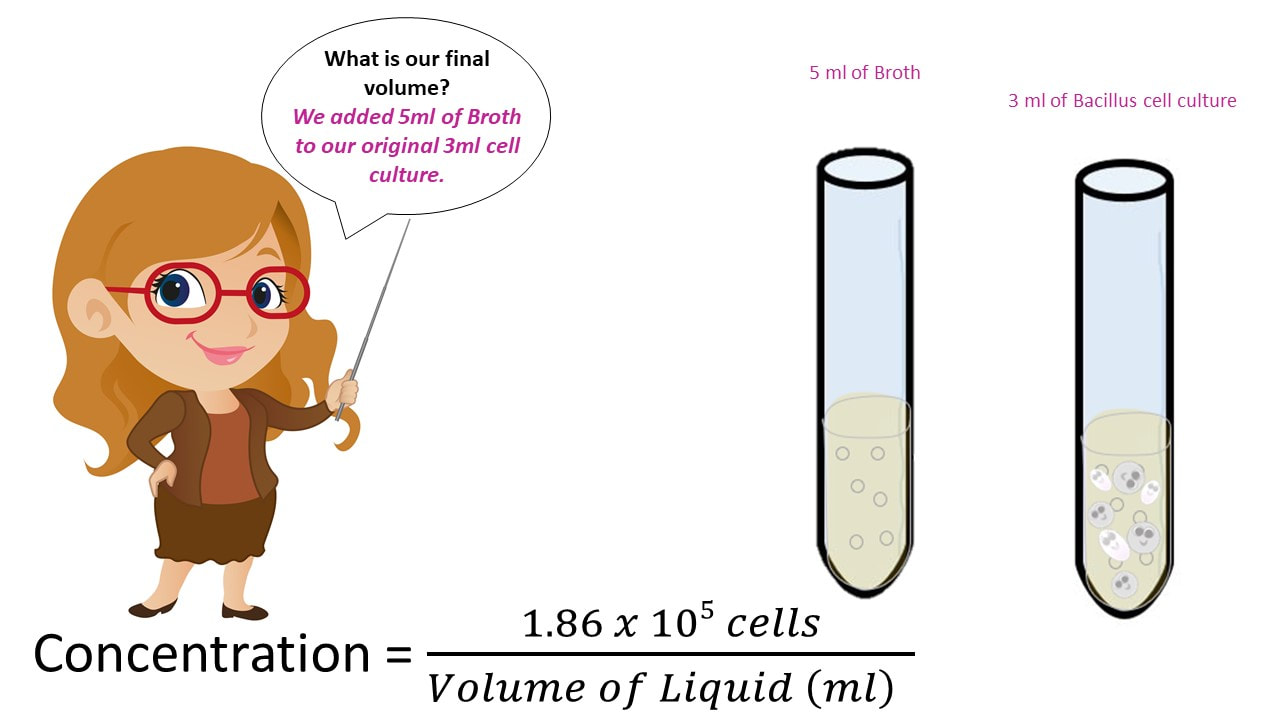
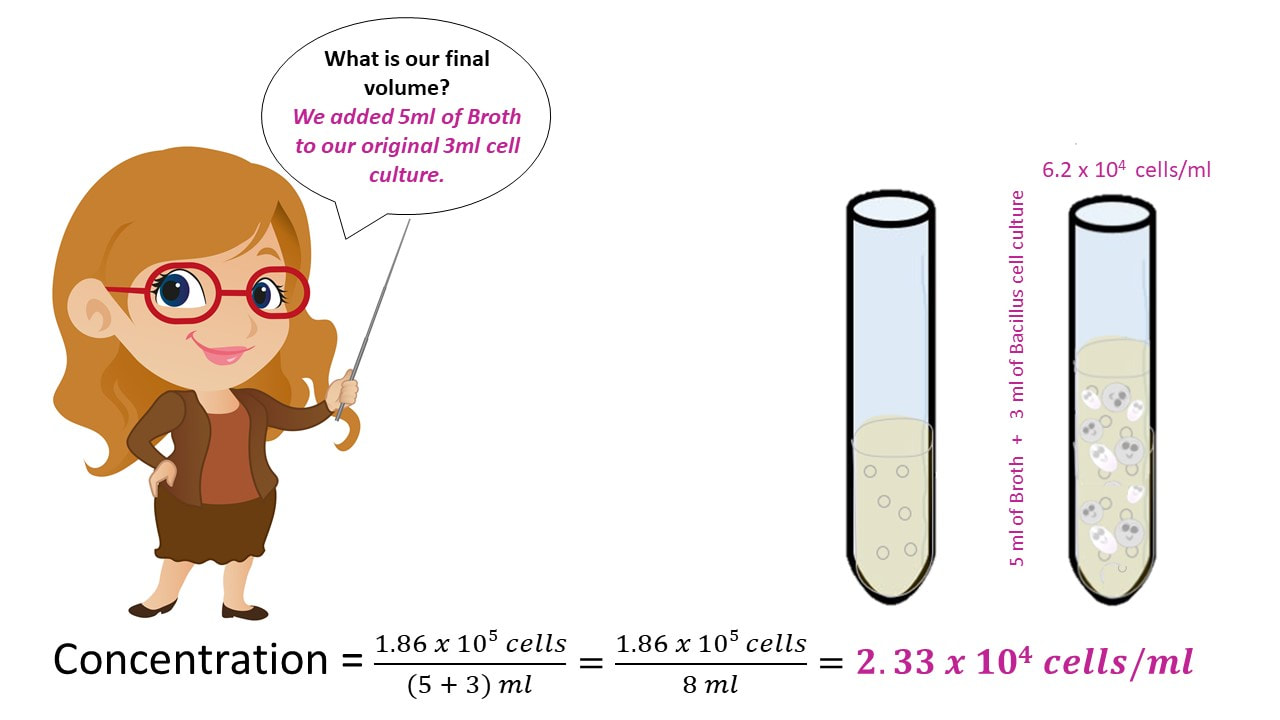
Let’s say I have 3 ml of a Bacillus cell culture with a concentration of 620,000 cells/ml. If I add 5 ml of TSB (nutrient broth) to the cell culture, what is my final concentration?

|
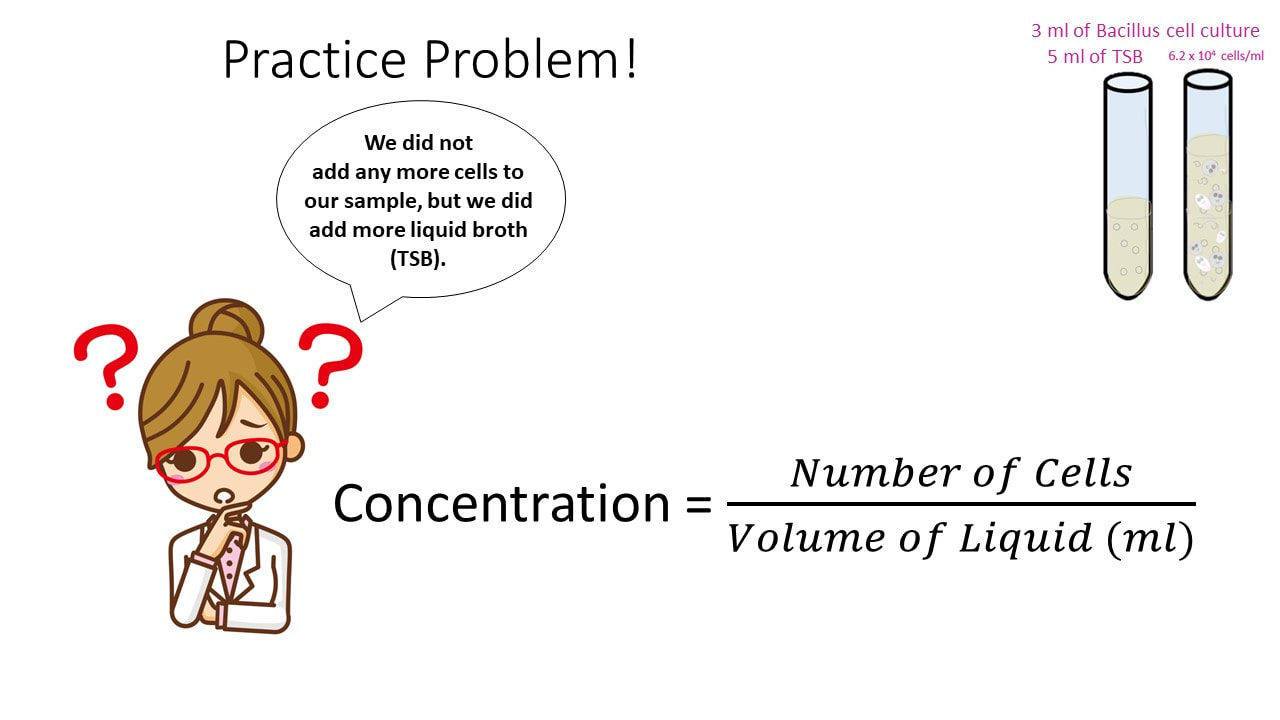
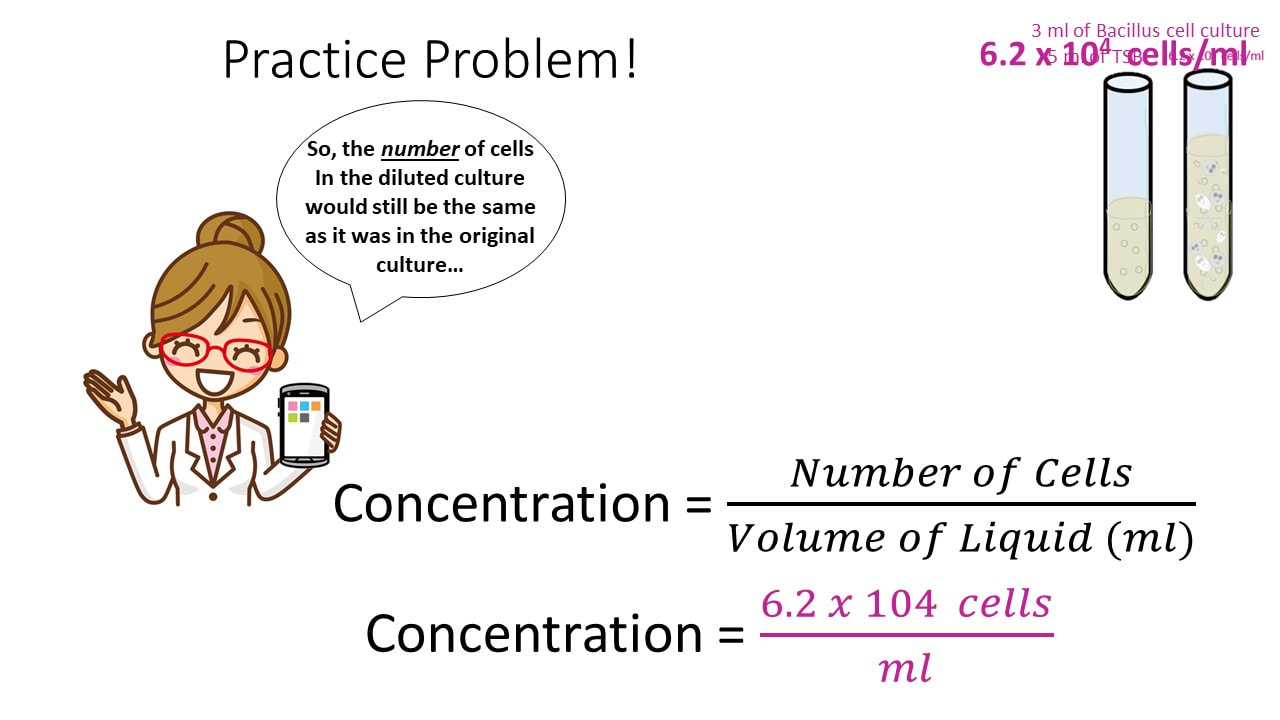


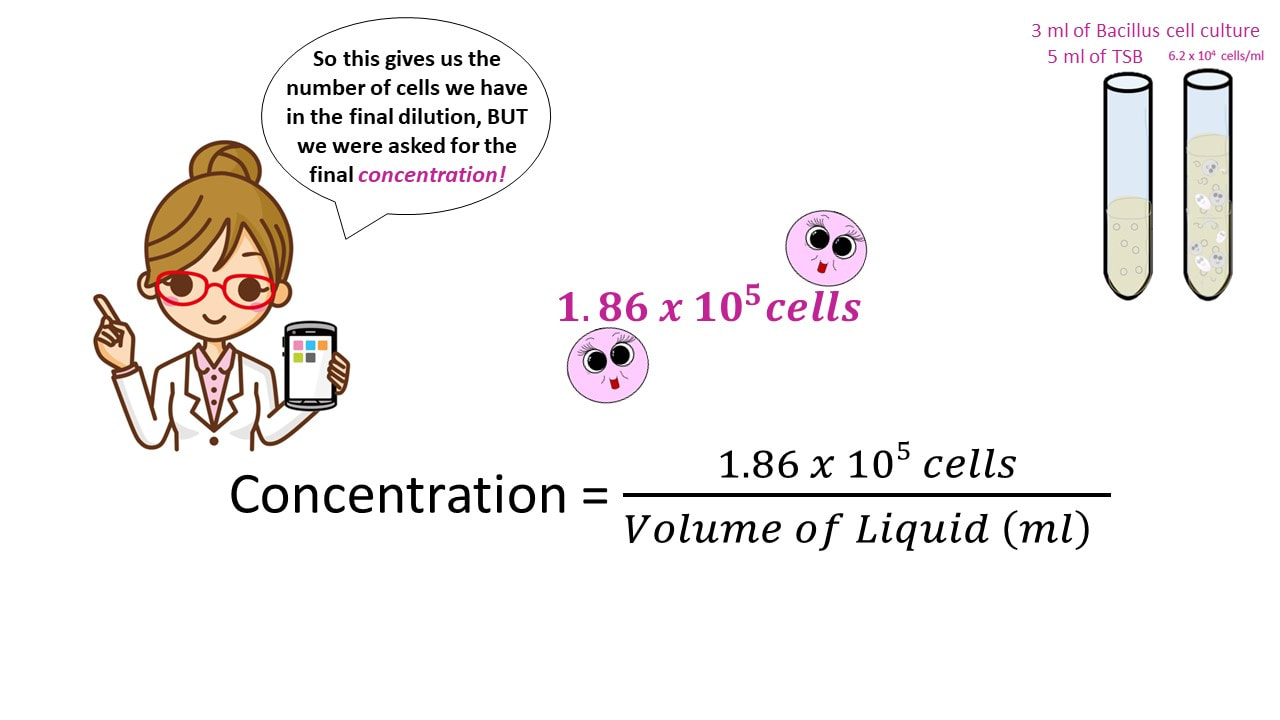
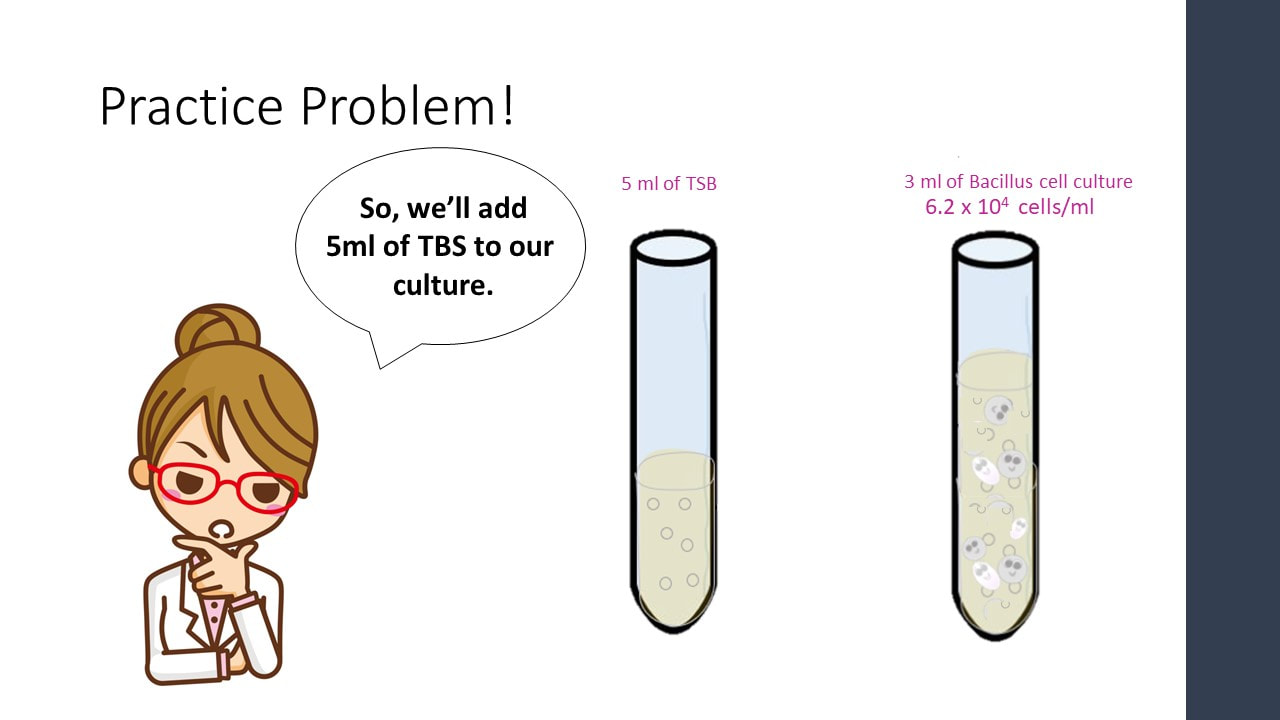
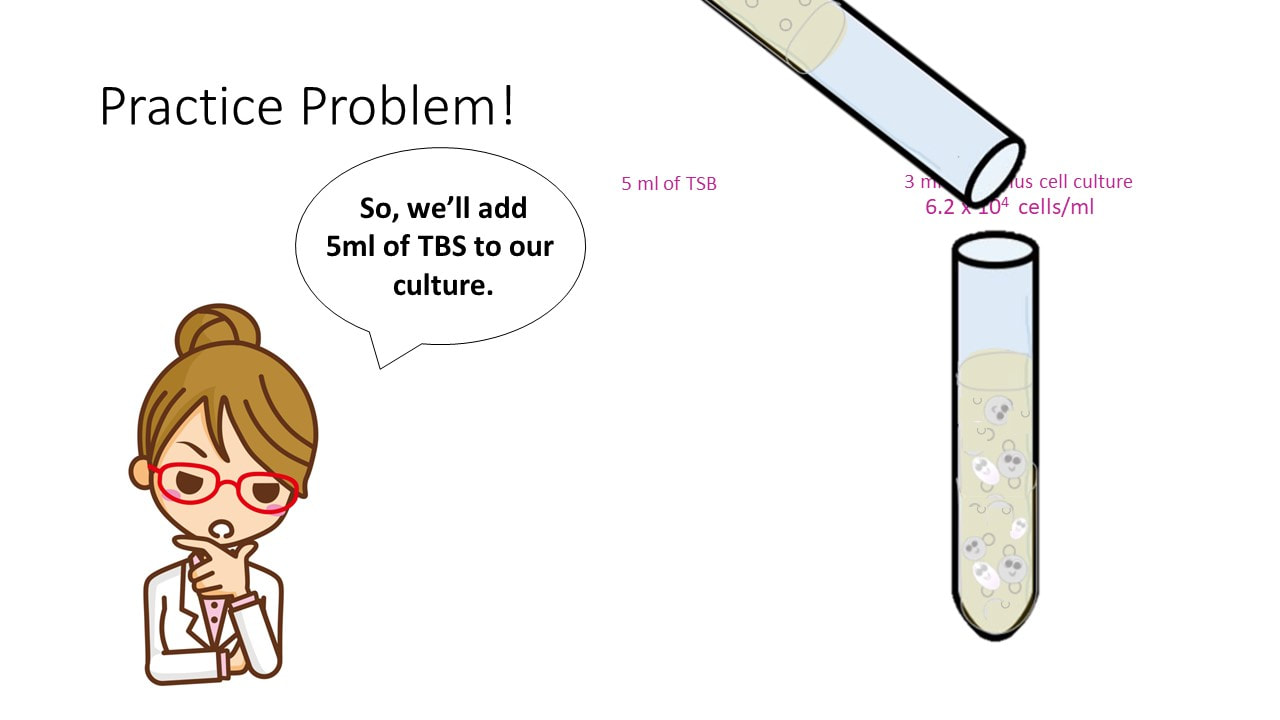
Concentration Practice Problem #3
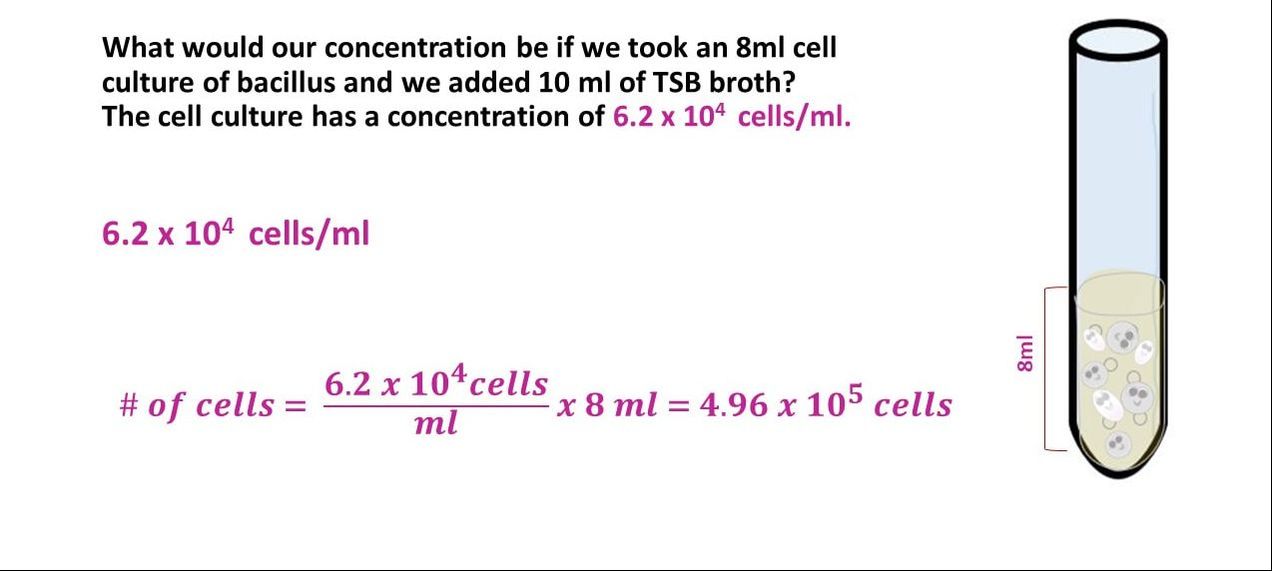
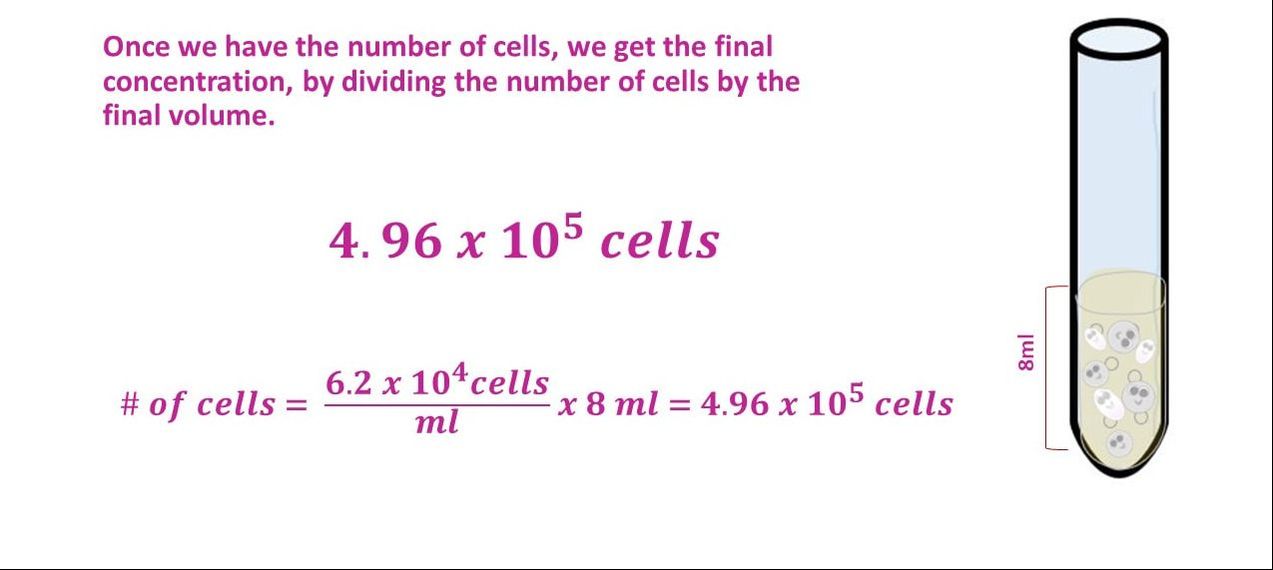
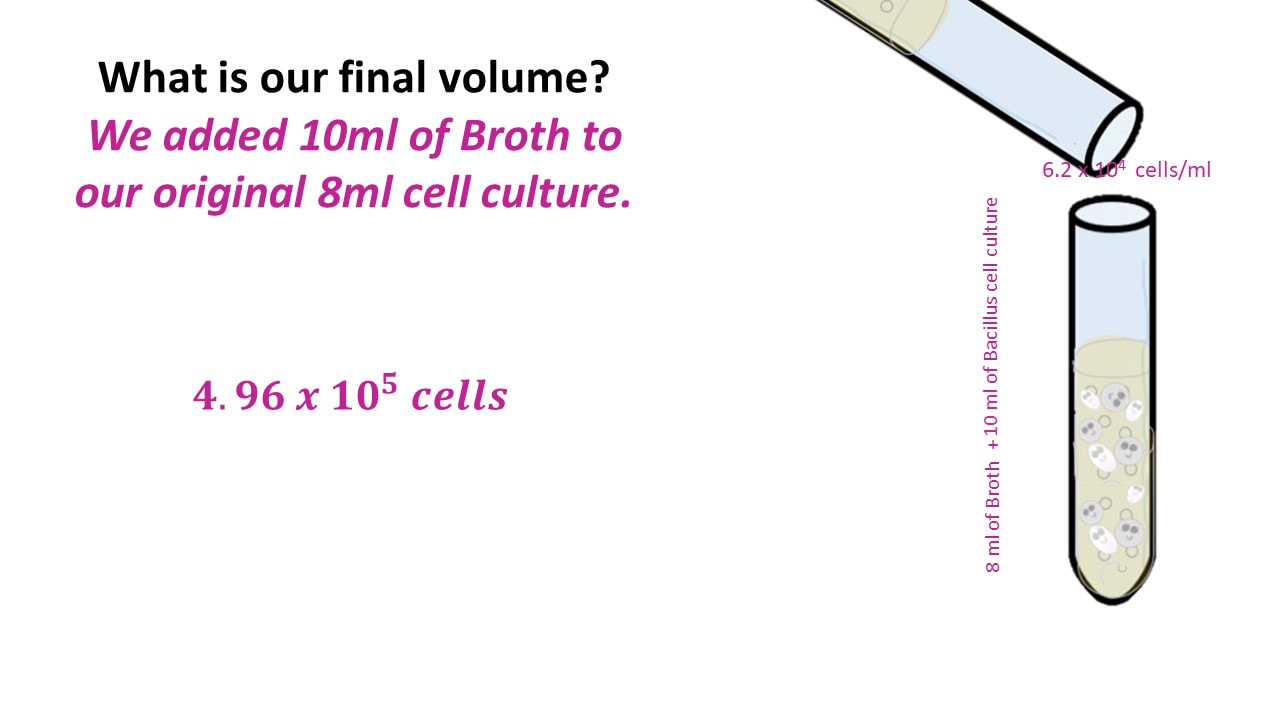
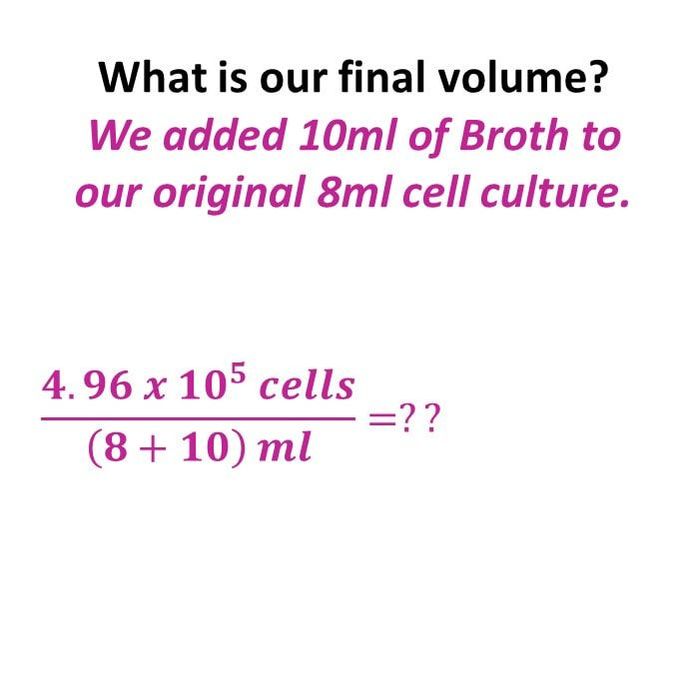
What would our concentration be if we took an 8 ml cell culture of bacillus and we added 10 ml of TSB broth? The cell culture has a concentration of 6.2 x 10^4 cells/ml.
Once we have the number of cells, we get the final concentration, by dividing the number of cells by the final volume.
Once we have the number of cells, we get the final concentration, by dividing the number of cells by the final volume.
|

|
ENUMERATIONS
- What is "enumeration" in microbiology? - Enumeration of microorganisms (quantifying or counting the number of cells or organisms in a culture)
- What does "viable" mean in microbiology? - Viable means "living".
The most common procedure for the enumeration of bacteria (quantifying or counting the number of cells or organisms in a culture) is the viable plate count method.
In this method, serial dilutions of a sample or cell culture containing viable or living microorganisms are plated onto a suitable growth medium such as TSA (trypsin soy agar) in a Petri dish. The most dilute sample is used to create a pour plate. The pour plate is created by pouring the entire contents of the dilute culture along with warm agar, into Petri plates, and allowing the contents to solidify at room temperature.
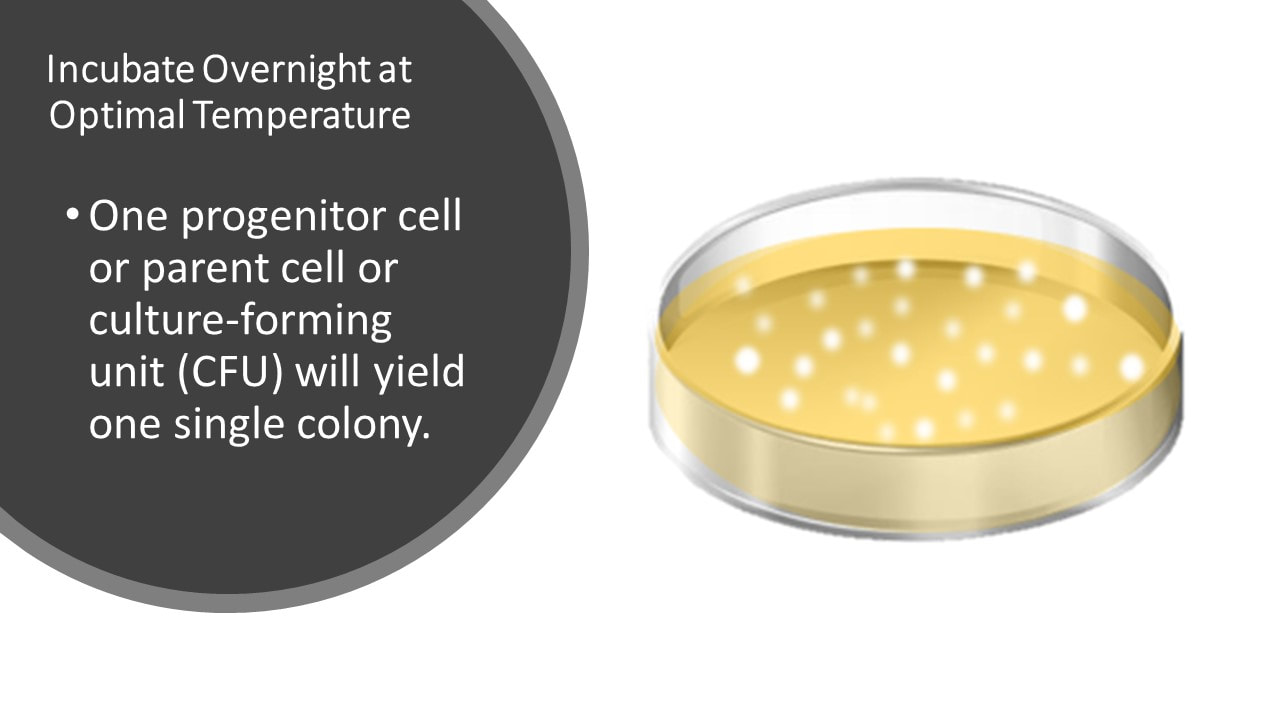
Once the pour plate has solidified, it can be incubated overnight at the optimal temperature for that organism.
It is assumed that each colony arises from an individual cell that has undergone cell division. Therefore, by counting the number of colonies and accounting for the dilution factor, the number of bacteria in the original sample can be determined.
Formula = (reciprocal of the dilution factor) x (# of cells or colonies) = # of cells in the original culture.
How do we count cells in culture?
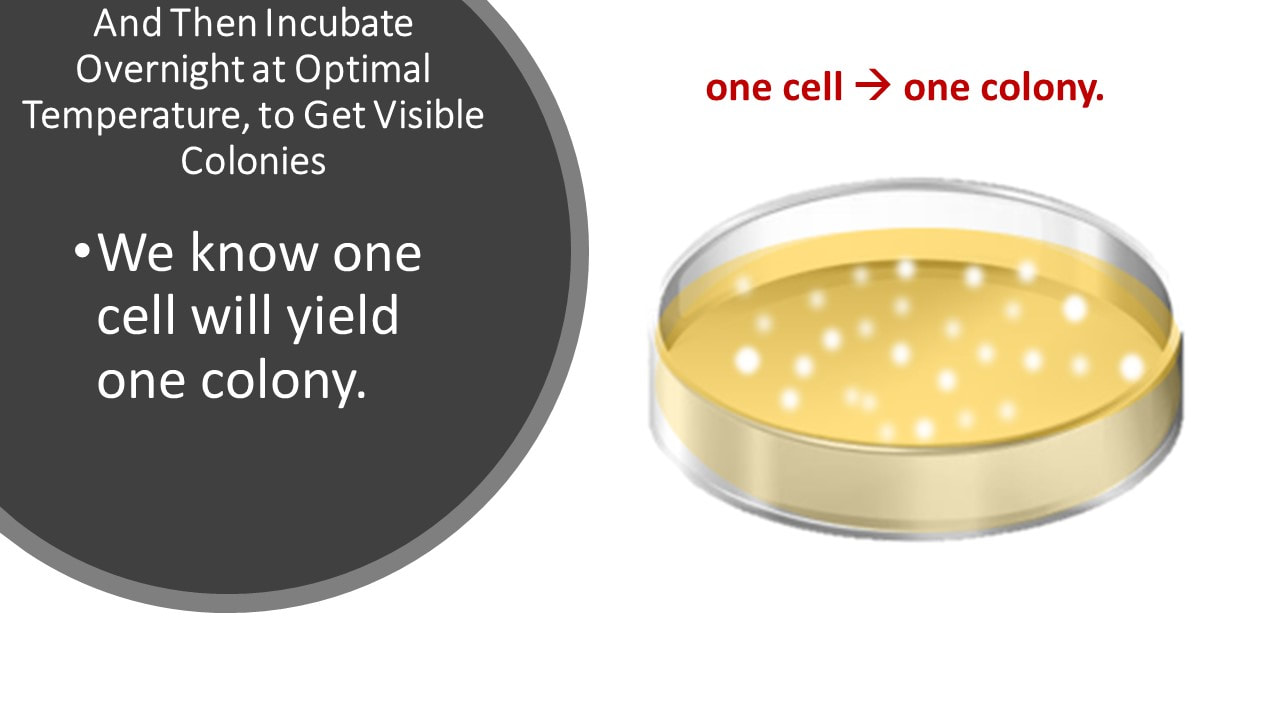
And Then Incubate Overnight at Optimal Temperature, to Get Visible Colonies
We know one cell will yield one colony.
We know one cell will yield one colony.

Calculate How Many Cells There Were in the Original Sample?


Calculate the Concentration of Cells in Original Sample.?

Practice Problem #4 - Why do we need to do a serial dilution in order to perform a viable plate count for enumeration?
BECAUSE there can be millions of cells in a single drop! So, doing the viable plate count method without a serial dilution, will give you something more like…..

Practice Problem #5
You have a cell culture that arrived to you already diluted, but they did not tell you the concentration of cells or the number of cells in the cell culture. It is also a small amount, it is not even 1 ml! In fact it is only 0.5 ml. If you made a pour plate with the entire 0.5 ml of your sample and grew 300 colonies, calculate the concentration of cells you had in the culture.

Then We Incubate Overnight at Optimal Temperature, to Get Visible Colonies
If we counted 300 colonies, then that means that we had 300 cells in the 0.5ml we used to make the plate.
If we counted 300 colonies, then that means that we had 300 cells in the 0.5ml we used to make the plate.


concentration = 300 cells
|
= 150 cells/ml
|
We want to get a sample of our cell culture that is dilute enough to yield between 30 and 300 colonies on a Petri plate/dish.

Setting up a serial dilution!
1:10
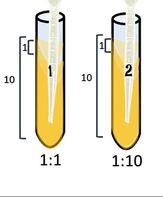
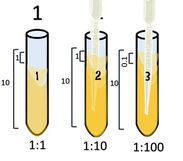


|
AND SO ON! |
It doesn't really matter how many dilutions you do, but you want to make sure you do enough dilutions and make enough culture plates so that you won't waste time having to repeat the experiment, in case your plates end up having too many colonies!



After you've made your serial dilutions, you will want to make some pour plates using a few of the most dilute concentrations from the series.
- Make sure to label your Petri dish with the dilution you used!
- Create Each Pour Plate Separately by Adding the Entire Contents of the Diluted Cell Culture to Warm TSA (Nutrient Agar) in a Petri Dish.
- Incubate Overnight at Optimal Temperature (You should check what the optimal temperature for growth is for your organism).
- Following the incubation, inspect the pour plates and discern which of the plates contains between 30 and 300 colonies. This is the plate we will want to use!
- Carefully count the colonies on the plate having between 30 and 300 colonies.
- Record the number of colonies on the plate
- Record the dilution factor that was used for that plate that was counted.
- Now, calculate the number of cells in the original 1 : 1 undiluted cell culture, by multiplying (the # of colonies you counted) by (the reciprocal dilution factor).
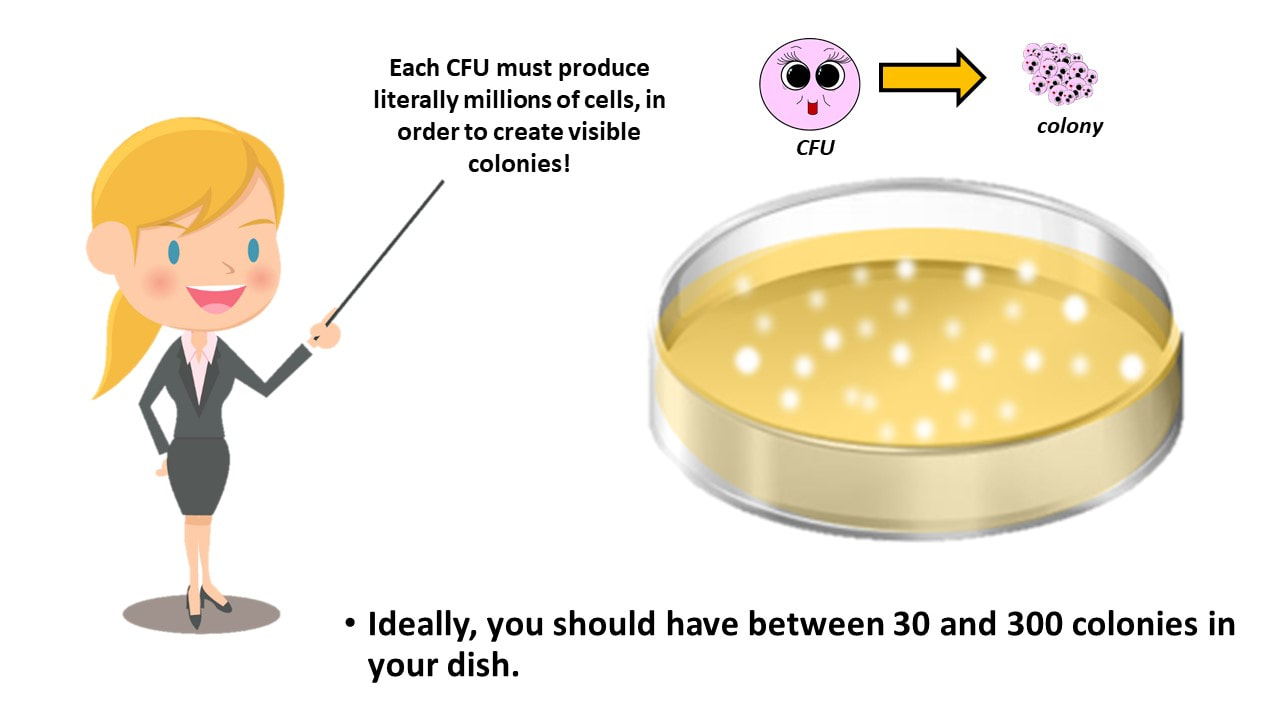


- Here's what's going on. One progenitor cell or parent cell or culture-forming unit (CFU) will yield one single colony. So counting the colonies that grew on our plate, gives us our cell count for our diluted cell culture.
We know what our dilution factor was that we used to make the plate.
Calculate the Number of Cells in Original Sample.
What is the concentration of a cell culture that grew 79 colonies on a Petri dish at a dilution of 1 : 100?

What is the concentration of a cell culture that was able to grow 28 colonies using only 0.1 ml of a 1:10,000 dilution sample?



HOW TO SET UP A 3-FOLD DILUTION!
Setting up a serial dilution!
1:3

|
AND SO ON! |
How to SET UP Serial Dilutions
A 1:2 dilution = 1/2 = a two-fold dilution
- Tube 1 must have 2 ml (equal to the denominator of the dilution factor, which is 2) of undiluted cells in nutrient broth (undiluted cell culture)
- All other tubes must have 1 ml total volume of nutrient broth only (these tubes must have 1 ml LESS than the denominator of the dilution factor, which is 2). This is so that when the 1 ml is transferred from one tube to the next each time, the total volume in the tube will be 2 ml (see video at start of page for animation!!)
- No matter what dilution factor we have, we will always transfer 1ml! This allows us to keep the math much easier to work with.


A 1 to 10 dilution series

A 1 to 5 dilution series
A 1:7 dilution = 1/7 = a seven-fold dilution
- Tube 1 must have 7 ml (equal to the denominator of the dilution factor, which is 7) of undiluted cells in nutrient broth (undiluted cell culture)
- All other tubes must have 6 ml total volume of nutrient broth only (these tubes must have 1 ml LESS than the denominator of the dilution factor, which is 7). This is so that when the 1 ml is transferred from one tube to the next each time, the total volume in the tube will be 7 ml (see video at start of page for animation!!)
- No matter what dilution factor we have, we will always transfer 1ml! This allows us to keep the math much easier to work with.

Dilution Series and Calculations
First, learn the language
A solution has 2 components.
The Value of Concentration is given as…
Concentration =𝑨𝒎𝒐𝒖𝒏𝒕 𝒎𝒂𝒔𝒔 𝒐𝒓 # 𝒐𝒇 𝑺𝒐𝒍𝒖𝒕𝒆𝑽𝒐𝒍𝒖𝒎𝒆 𝒐𝒇 𝑺𝒐𝒍𝒗𝒆𝒏𝒕
First, learn the language
Concentration is a RATIO that expresses the amount of solids you have to the amount of liquid you have.
The Value of Concentration is given as…
Concentration=𝑨𝒎𝒐𝒖𝒏𝒕 𝒎𝒂𝒔𝒔 𝒐𝒓 # 𝒐𝒇 𝑺𝒐𝒍𝒖𝒕𝒆𝑽𝒐𝒍𝒖𝒎𝒆 𝒐𝒇 𝑺𝒐𝒍𝒗𝒆𝒏𝒕
What is Concentration?
Concentration = 𝑺𝒐𝒍𝒖𝒕𝒆𝑺𝒐𝒍𝒗𝒆𝒏𝒕 = # 𝒐𝒇 𝒄𝒆𝒍𝒍𝒔𝑽𝒐𝒍𝒖𝒎𝒆 𝒐𝒇 𝑩𝒓𝒐𝒕𝒉
For example, lets say you have 5 ml of broth containing
37 (or 10,000,000) cells.
You would have a concentration of…
# 𝑜𝑓 𝑐𝑒𝑙𝑙𝑠𝑉𝑜𝑙𝑢𝑚𝑒 𝑜𝑓 𝐵𝑟𝑜𝑡ℎ = 10,000,000 𝑐𝑒𝑙𝑙𝑠5 𝑚𝑙 𝑜𝑓 𝐵𝑟𝑜𝑡ℎ = which reduces to 2,000,000 𝑐𝑒𝑙𝑙𝑠1 𝑚𝐿 𝑜𝑓 𝐵𝑟𝑜𝑡ℎ =
Which can be written as 2,000,000 𝑐𝑒𝑙𝑙𝑠𝑚𝑙 or 2 x 36 𝑐𝑒𝑙𝑙𝑠𝑚𝑙
Dilution Problems
A “two-fold” dilution = a 1:2 dilution (whenever a number is written in the form 1:2, this is a ratio and is simply another way to write 12)
Concentration = 𝑆𝑜𝑙𝑢𝑡𝑒𝑆𝑜𝑙𝑣𝑒𝑛𝑡 à 𝑁𝑢𝑚𝑏𝑒𝑟 𝑜𝑓 𝐶𝑒𝑙𝑙𝑠𝑉𝑜𝑙𝑢𝑚𝑒 𝑜𝑓 𝑆𝑜𝑙𝑣𝑒𝑛𝑡 x (12)
Scientific Notation
In microbiology, it is convenient to use scientific notation, because we are often dealing with huge numbers of cells.
Scientific Notation
This works for small numbers too!
We just count the number of zeros that come AFTER the decimal point, and use a negative sign for our exponent.
Practice Problem!
Let’s say we have a Bacillus cell culture with a concentration of 6.2 x 104 cells/ml.
If I have 3 ml of the Bacillus cell culture and add 5 ml of TSB (nutrient broth) to the cell culture, what is my final concentration?
Practice Problem!
Practice Problem!
Practice Problem!
Practice Problem!
Practice Problem!
Practice Problem!
Practice Problem!
Practice Problem #2
What would our concentration be if we took an 8ml cell culture of bacillus and we added 3 ml of TSB broth? The cell culture has a concentration of 6.2 x 104 cells/ml.
Practice Problem #2
Once we have the number of cells, we get the final concentration, by dividing the number of cells by the final volume.
Practice Problem!
Practice Problem!
How do we count cells in culture?
The cells are way to small to just simply count directly!
If the culture is dilute enough, we could just mix our entire sample with warm agar...
And Then Incubate Overnight at Optimal Temperature, to Get Visible Colonies
We know one cell will yield one colony.
How Many Cells Were in the Original Sample?
How Many Cells Were in the Original Sample?
How Many Cells Were in the Original Sample?
Calculate the Concentration of Cells in Original Sample.
And Then Incubate Overnight at Optimal Temperature, to Get Visible Colonies
We know one cell will yield one colony.
BUT…. Here’s the problem! There can be millions of cells in a single drop! So, doing this is the without a serial dilution, will give you something more like…..
All of our previous ideas will work, once we get a sample of our cell culture that is dilute enough to yield between 30 and 300 colonies on a Petri plate/dish.
What if we did this same idea again, and just kept track of the volume?
We’ll Use Numbers that are Super Easy to Work With!
Setting up a serial dilution!
1:10
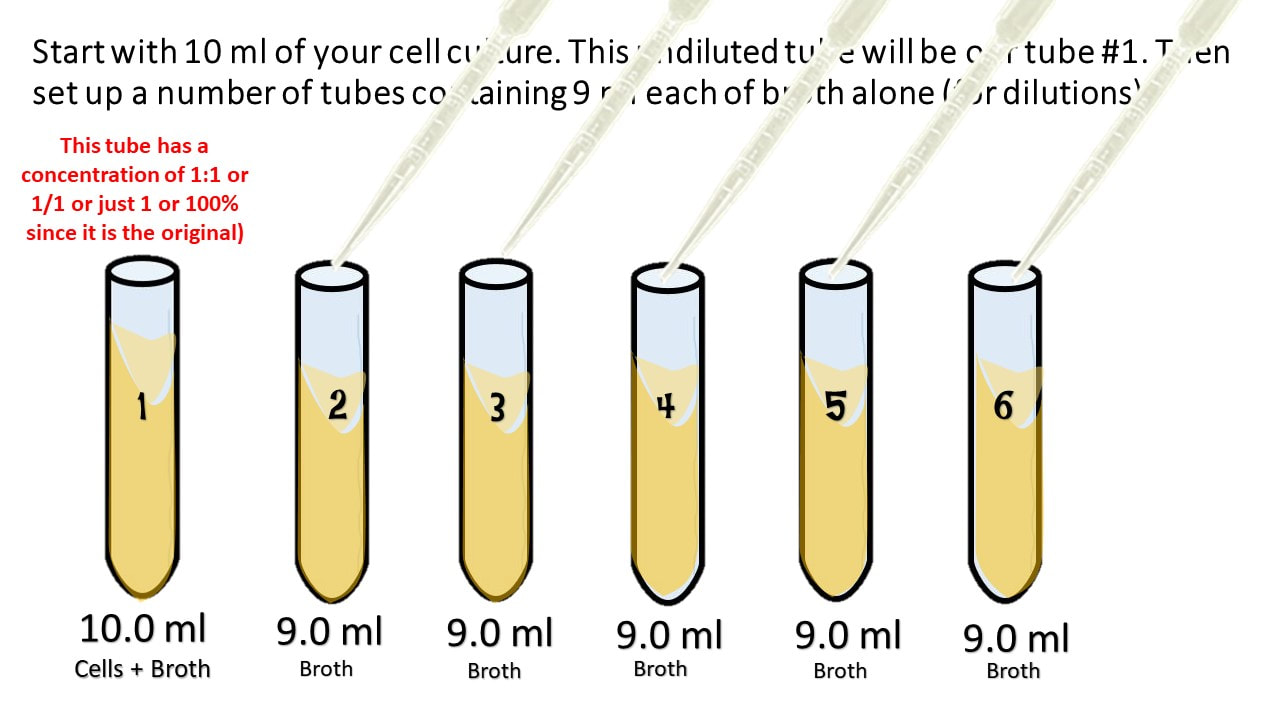
Start with 10 ml of your cell culture. This undiluted tube will be our tube #1. Then set up a number of tubes containing 9 ml each of broth alone (for dilutions).
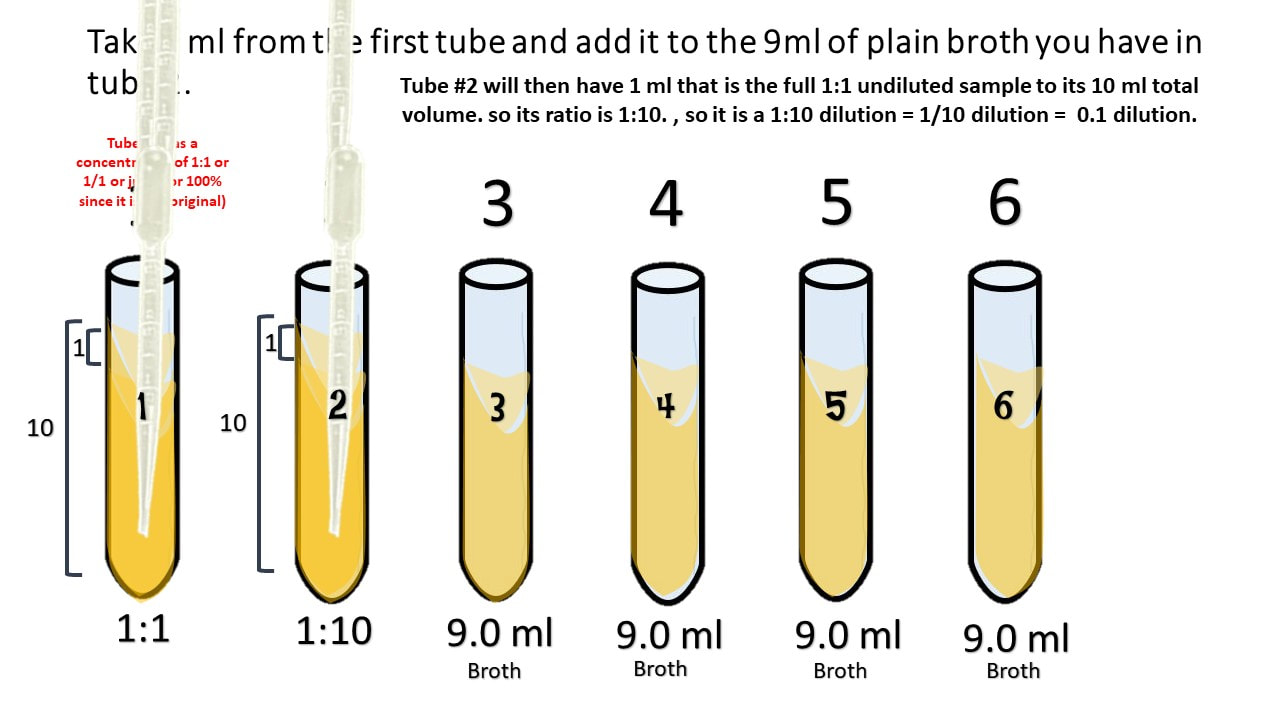
Take 1 ml from the first tube and add it to the 9ml of plain broth you have in tube 2.
Take 1 ml from tube #2 and [place it into tube #3.
Tube #3 has 1 part that is from the 1:10 dilution, to its 10 ml total volume. So its ratio is 0.1/10 which reduces to 1/100. which = 0.01.
Take 1 ml from tube #3 and place it into tube #4.
Tube #4 has 1 ml from 1:100 which is 0.01 to its total volume of 10 ml, so its ratio is 0.01:10 = 𝟎.𝟎𝟏𝟏𝟎= 1:1000.
Take 1 ml from tube #4 and place it into tube #5.
Tube #5 has 1 ml from 1:1000 which is 0.001 to its total volume of 10 ml, so its ratio is 0.001:10 = 𝟎.𝟎𝟎𝟏𝟏𝟎= 1:10,000.
Take 1 ml from tube #5 and place it into tube #6.
Tube #6 has 1 ml from 1:10,000 which is 0.0001 to its total volume of 10 ml, so its ratio is 0.0001:10 = 𝟎.𝟎𝟎𝟎𝟏𝟏𝟎= 1:100,000.
Make a Pour Plate Cell Culture by Adding the Entire Contents of the Most Dilute Tube (Tube #6) by Adding it to Warm Agar in a Petri Dish.
Make sure to label your Petri dish with the dilution you used!
You should also make a culture of tube #5 and #4, just to be safe!
Incubate Overnight at Optimal Temperature
One progenitor cell or parent cell or culture-forming unit (CFU) will yield one single colony.
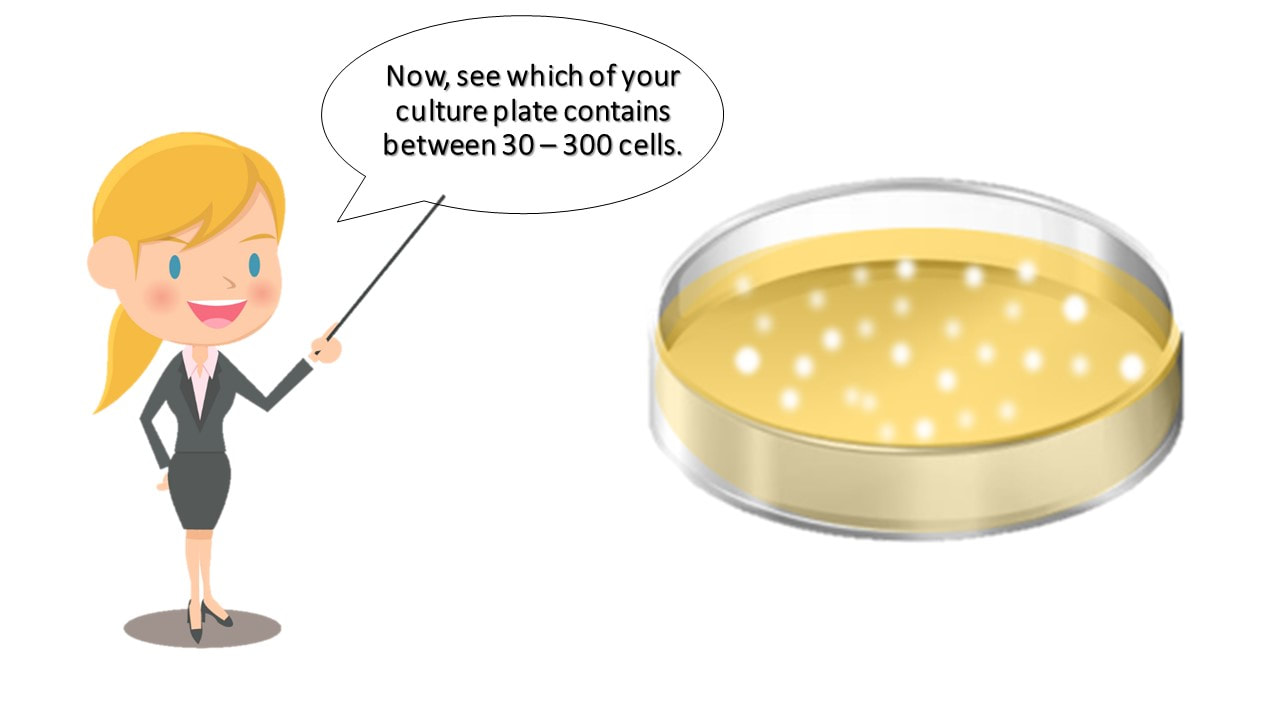
Now, see which of your culture plate contains between 30 – 300 colonies.
Ideally, you should have between 30 and 300 colonies in your dish.
Ideally, you should have one of your plates contain between 30 and 300 colonies.
COUNT THE COLONIES
# COLONIES = Count the bacterial colonies on the plate that grew between 30 and 300 colonies.
THE RECIPROCAL OF THE DILUTION FACTOR à
The Dilution Factor used for the plate with the colonies we counted,
= 𝟏𝟏𝟎,𝟎𝟎𝟎
To get the reciprocal, simply flip the fraction upside-down
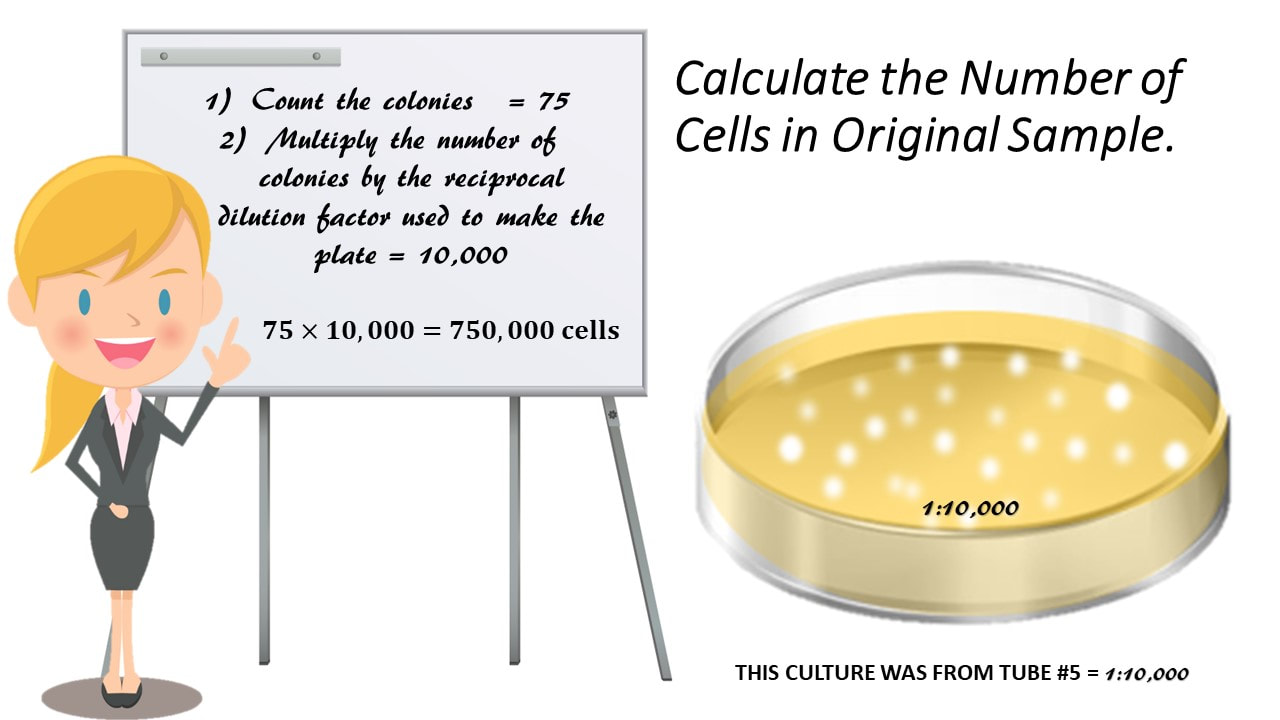
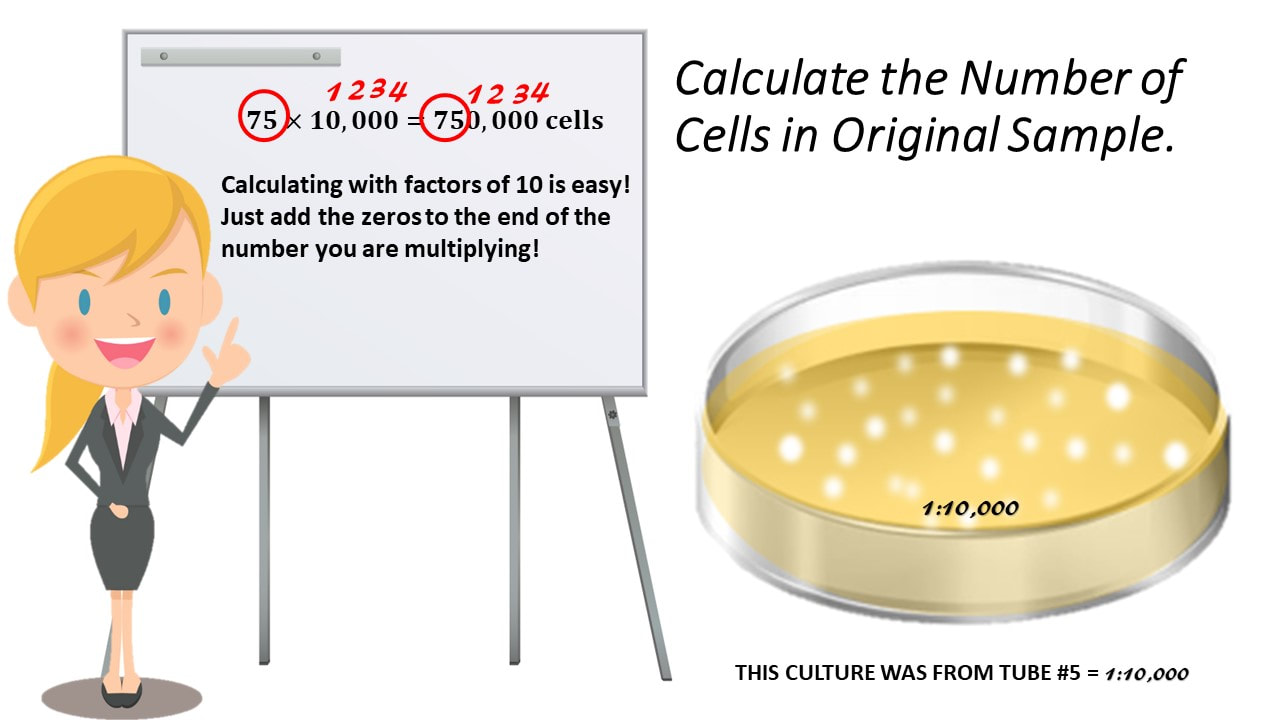
= 𝟏𝟎,𝟎𝟎𝟎𝟏 = 10,000
Calculate the Number of Cells in Original Sample.
Calculate the Number of Cells in Original Sample.
Practice Problem #1
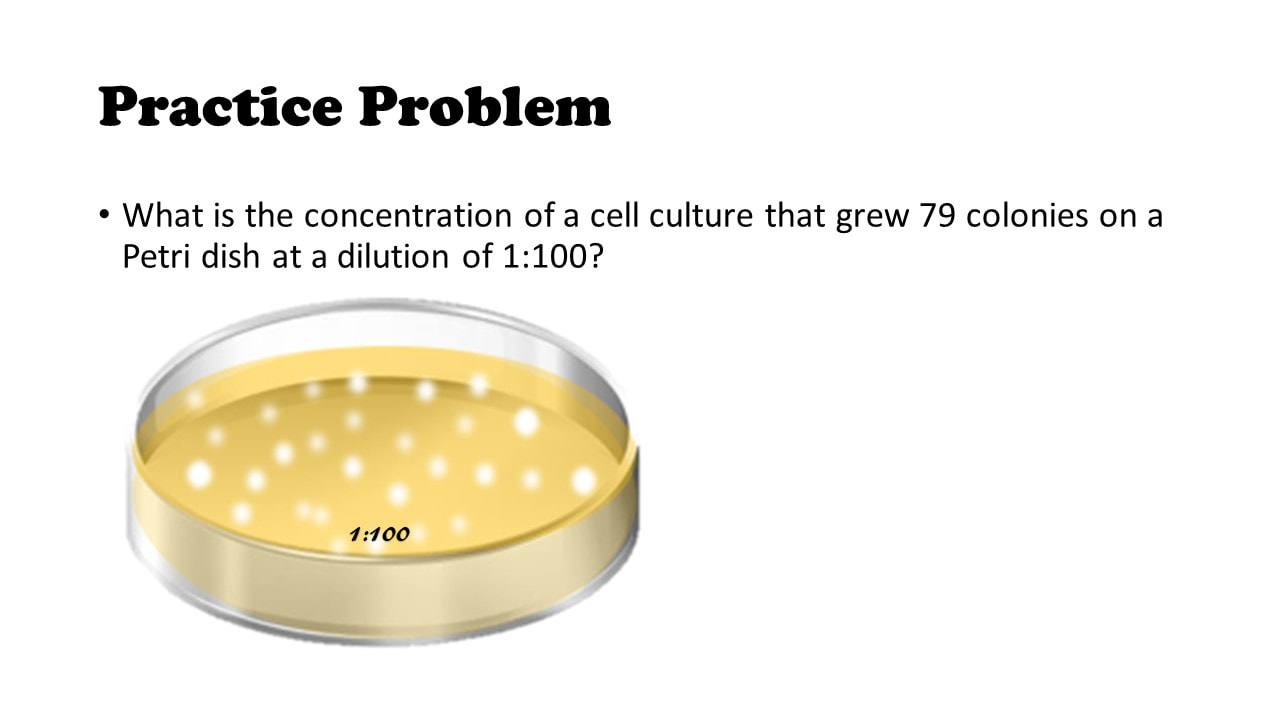
What is the concentration of a cell culture that grew 79 colonies on a Petri dish at a dilution of 1:100?
Practice Problem #1
What is the concentration of a cell culture that grew 79 colonies on a Petri dish at a dilution of 1:100?
Practice Problem #2
How many cell did we start with in our original cell culture?
Practice Problem #2
What is the concentration of a cell culture that grew 79 colonies on a Petri dish at a dilution of 1:100?
Calculate the Number of Cells in Original Sample.
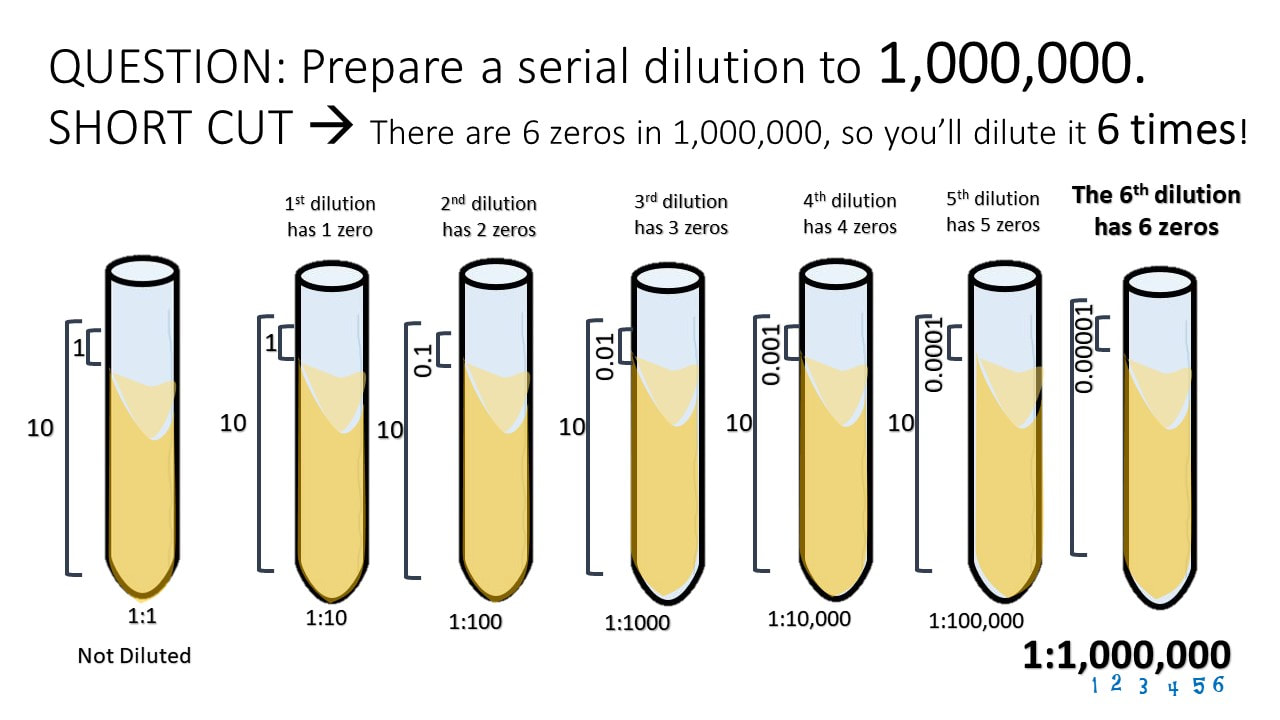
QUESTION: Prepare a serial dilution to 1,000,000. SHORT CUT à There are 6 zeros in 1,000,000, so you’ll dilute it 6 times!
MATH CHEAT SHEET!
100 = 1 101 = 10 102 = 100 103 = 1000 104 = 10,000 105 = 100,000
106 = 1,000,000 107 = 10,000,000 108 = 100,000,000 109 = 1,000,000,000 1010= 10,000,000,000
100
MATH CHEAT SHEET!
10-1 = 0.1 = 1/10
10-2 = 0.01 = 1/100
10-3 = 0.001 = 1/1000
10-4 = 0.0001 = 1/10000
10-5 = 0.00001 = 1/100000
10-6 = 0.000001 = 1/1000000
10-7 = 0.0000001 = 1/10000000
10-8 = 0.00000001 = 1/100000000
10-9 = 0.000000001 = 1/1000000000
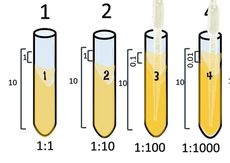
HOW TO SET UP A 3-FOLD DILUTION!
Setting up a serial dilution!
1:3
Start with 3 ml of your cell culture. This undiluted tube will be our tube #1. Then set up a number of tubes containing 2 ml each of broth alone (for dilutions).
Take 1 ml from the first tube and add it to the 2ml of plain broth you have in tube 2.
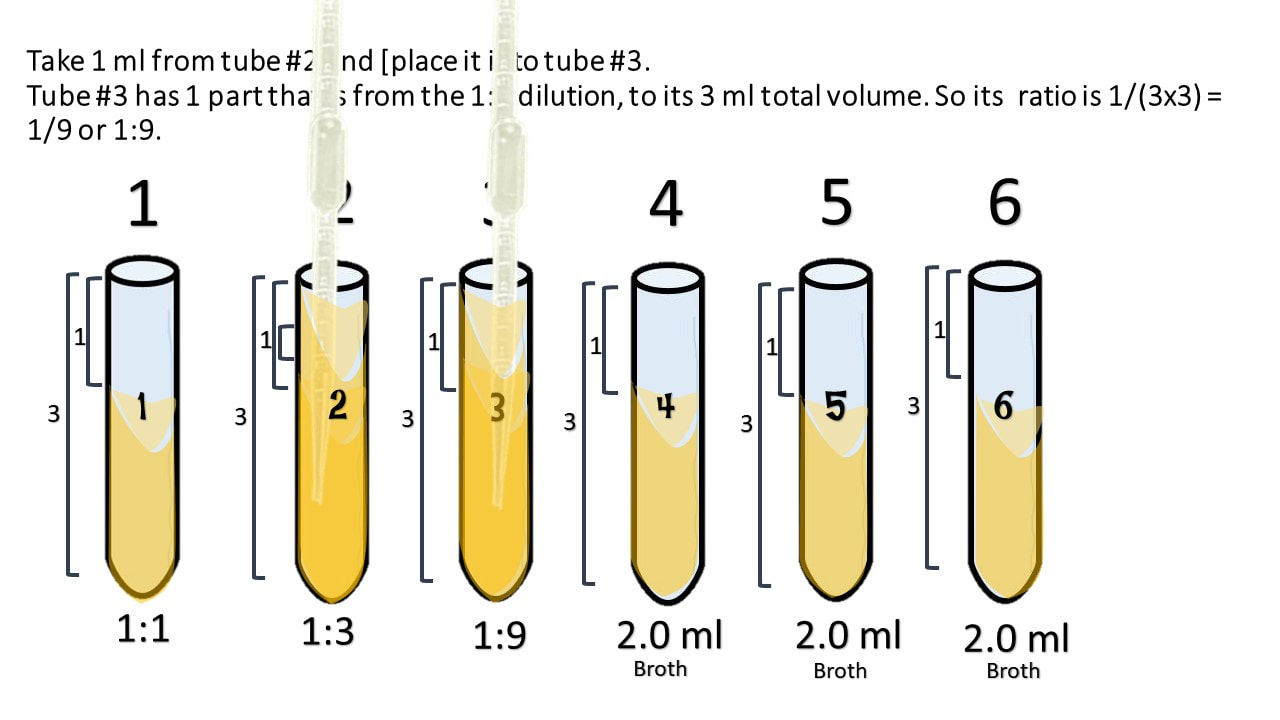
Take 1 ml from tube #2 and [place it into tube #3.
Tube #3 has 1 part that is from the 1:3 dilution, to its 3 ml total volume. So its ratio is 1/(3x3) = 1/9 or 1:9.
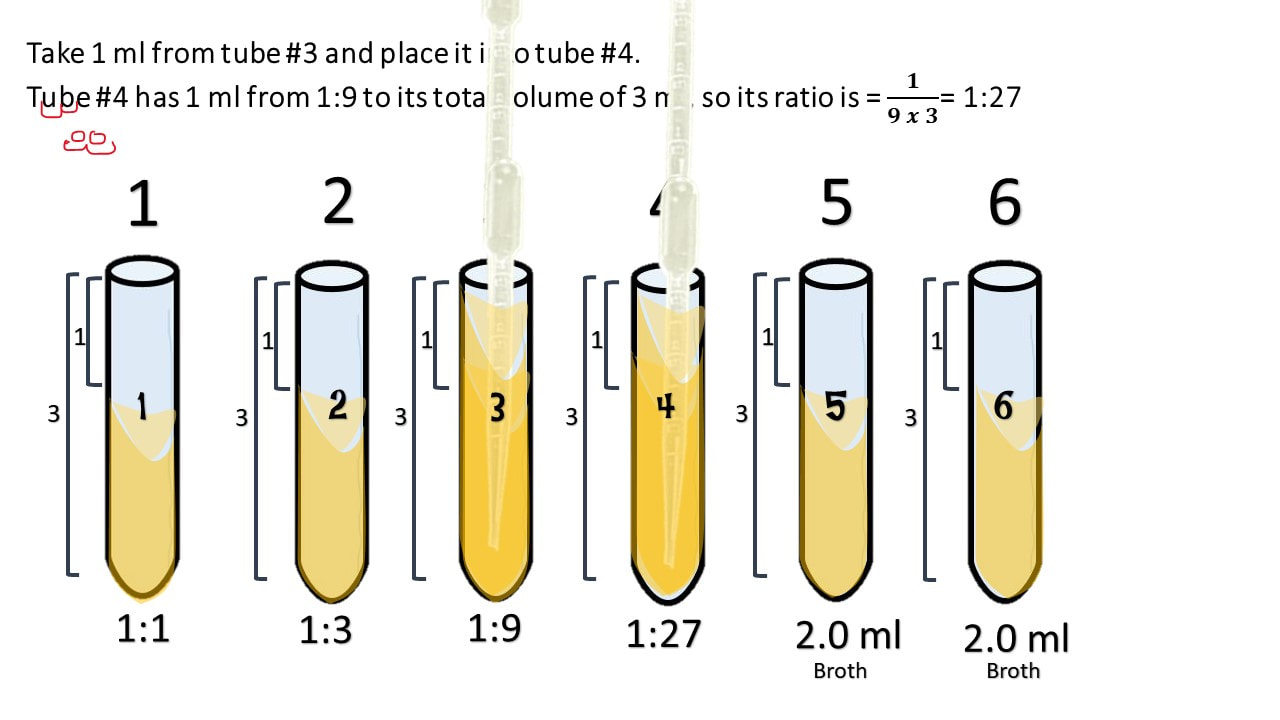
Take 1 ml from tube #3 and place it into tube #4.
Tube #4 has 1 ml from 1:9 to its total volume of 3 ml, so its ratio is = 𝟏𝟗 𝒙 𝟑= 1:27
Take 1 ml from tube #4 and place it into tube #5.
Tube #5 has 1 ml from 1:27 to its total volume of 3 ml, so its ratio is = 𝟏𝟐𝟕 𝒙 𝟑= 1:81
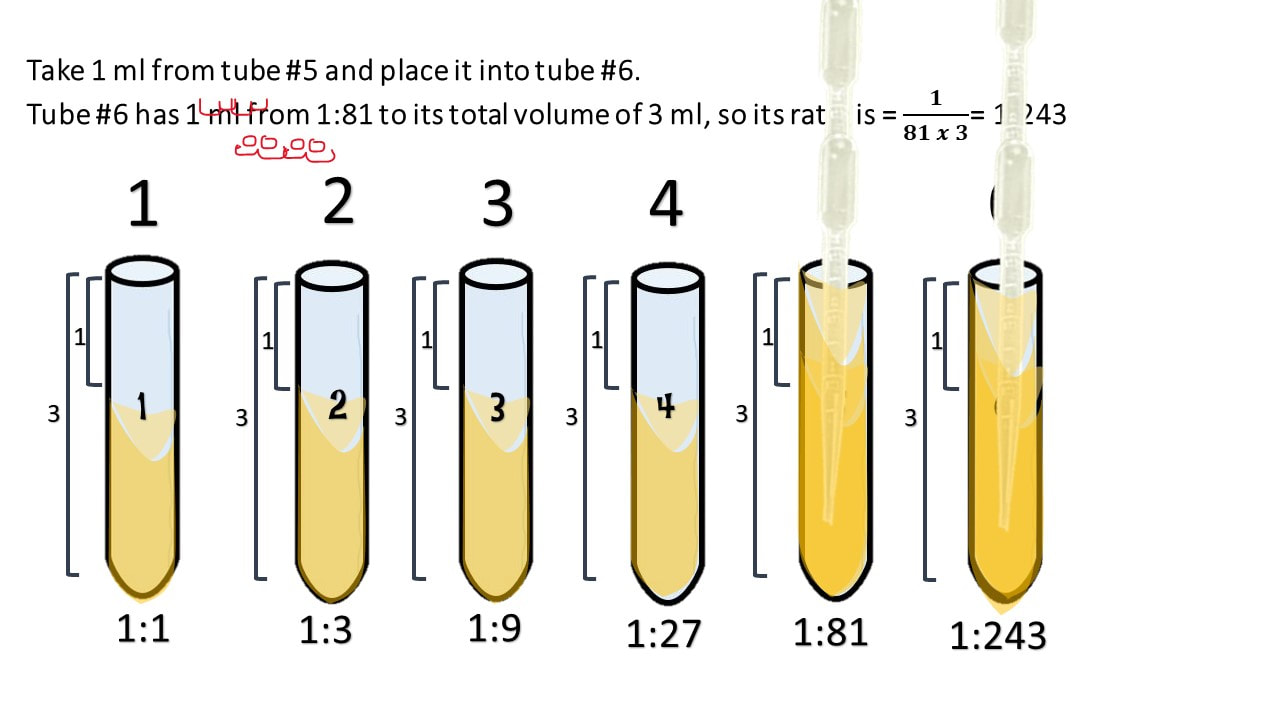
Take 1 ml from tube #5 and place it into tube #6.
Tube #6 has 1 ml from 1:81 to its total volume of 3 ml, so its ratio is = 𝟏𝟖𝟏 𝒙 𝟑= 1:243
Make a Pour Plate Cell Culture by Adding the Entire Contents of the Most Dilute Tube (Tube #6) by Adding it to Warm Agar in a Petri Dish.
Make sure to label your Petri dish with the dilution you used!
You should also make a culture of tube #5 and #4, just to be safe!
Incubate Overnight at Optimal Temperature
One progenitor cell or parent cell or culture-forming unit (CFU) will yield one single colony.
Now, see which of your culture plate contains between 30 – 300 cells.
Ideally, you should have between 30 and 300 colonies in your dish.
Ideally, you should have one of your plates contain between 30 and 300 colonies.
COUNT THE COLONIES
# COLONIES = Count the bacterial colonies on the plate that grew between 30 and 300 colonies.
THE RECIPROCAL OF THE DILUTION FACTOR à
The Dilution Factor used for the plate with the colonies we counted,
To get the reciprocal, simply flip the fraction upside-down
Calculate the Number of Cells in Original Sample.
1:10 Dilution Series
1:5 Dilution Series
1:2 Dilution Series
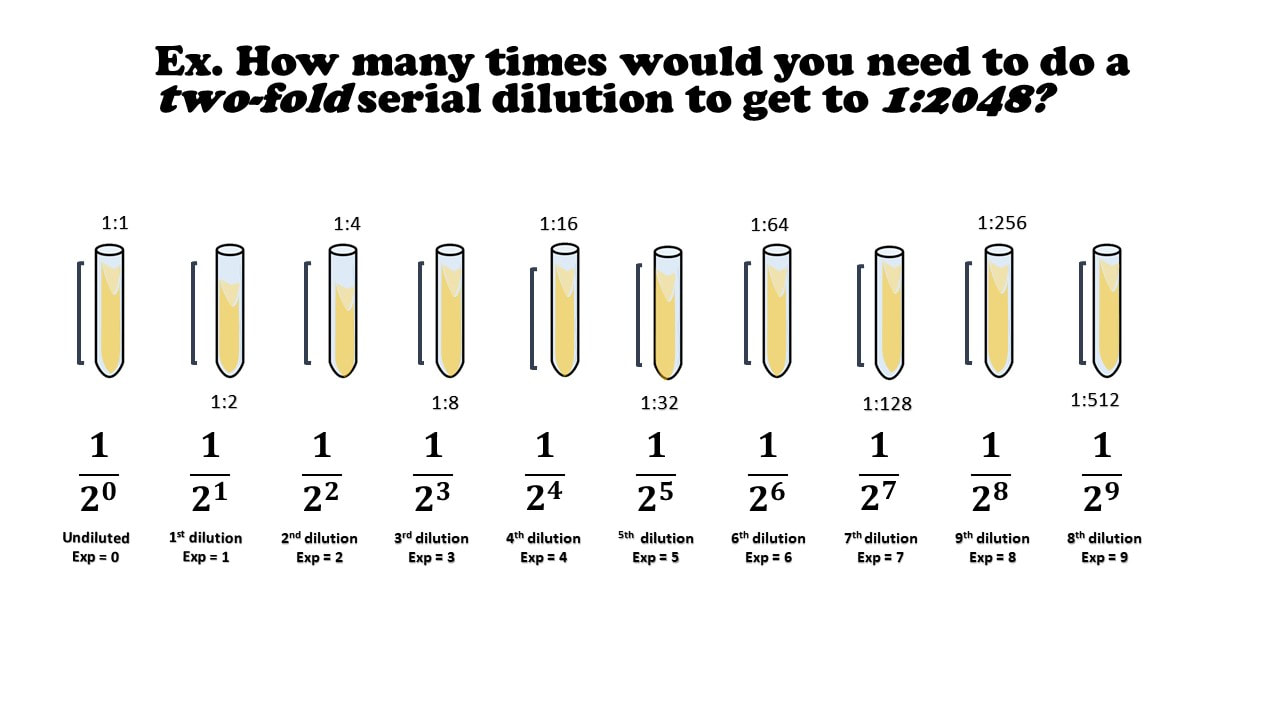
Ex. How many times would you need to do a two-fold serial dilution to get to 1:2048?
There’s an even faster way, but you’ll need a calculator.
Ex. How many times would you need to do a two-fold serial dilution to get to 1:2048?
Now go
CELL-ebrate your new skills!
THANK YOU FOR WATCHING!
Created by ScientistCindy.com
First, learn the language
A solution has 2 components.
- Solute – the solid component of the solution à In our example, our solid components our the cells!
- Solvent (Diluent) – the liquid component of the solution à In our example, our solvent is nutrient broth.
The Value of Concentration is given as…
Concentration =𝑨𝒎𝒐𝒖𝒏𝒕 𝒎𝒂𝒔𝒔 𝒐𝒓 # 𝒐𝒇 𝑺𝒐𝒍𝒖𝒕𝒆𝑽𝒐𝒍𝒖𝒎𝒆 𝒐𝒇 𝑺𝒐𝒍𝒗𝒆𝒏𝒕
First, learn the language
Concentration is a RATIO that expresses the amount of solids you have to the amount of liquid you have.
The Value of Concentration is given as…
Concentration=𝑨𝒎𝒐𝒖𝒏𝒕 𝒎𝒂𝒔𝒔 𝒐𝒓 # 𝒐𝒇 𝑺𝒐𝒍𝒖𝒕𝒆𝑽𝒐𝒍𝒖𝒎𝒆 𝒐𝒇 𝑺𝒐𝒍𝒗𝒆𝒏𝒕
What is Concentration?
Concentration = 𝑺𝒐𝒍𝒖𝒕𝒆𝑺𝒐𝒍𝒗𝒆𝒏𝒕 = # 𝒐𝒇 𝒄𝒆𝒍𝒍𝒔𝑽𝒐𝒍𝒖𝒎𝒆 𝒐𝒇 𝑩𝒓𝒐𝒕𝒉
For example, lets say you have 5 ml of broth containing
37 (or 10,000,000) cells.
You would have a concentration of…
# 𝑜𝑓 𝑐𝑒𝑙𝑙𝑠𝑉𝑜𝑙𝑢𝑚𝑒 𝑜𝑓 𝐵𝑟𝑜𝑡ℎ = 10,000,000 𝑐𝑒𝑙𝑙𝑠5 𝑚𝑙 𝑜𝑓 𝐵𝑟𝑜𝑡ℎ = which reduces to 2,000,000 𝑐𝑒𝑙𝑙𝑠1 𝑚𝐿 𝑜𝑓 𝐵𝑟𝑜𝑡ℎ =
Which can be written as 2,000,000 𝑐𝑒𝑙𝑙𝑠𝑚𝑙 or 2 x 36 𝑐𝑒𝑙𝑙𝑠𝑚𝑙
Dilution Problems
A “two-fold” dilution = a 1:2 dilution (whenever a number is written in the form 1:2, this is a ratio and is simply another way to write 12)
Concentration = 𝑆𝑜𝑙𝑢𝑡𝑒𝑆𝑜𝑙𝑣𝑒𝑛𝑡 à 𝑁𝑢𝑚𝑏𝑒𝑟 𝑜𝑓 𝐶𝑒𝑙𝑙𝑠𝑉𝑜𝑙𝑢𝑚𝑒 𝑜𝑓 𝑆𝑜𝑙𝑣𝑒𝑛𝑡 x (12)
Scientific Notation
In microbiology, it is convenient to use scientific notation, because we are often dealing with huge numbers of cells.
Scientific Notation
- Round the number so you only have 3 “non-zero” digits. This is a good rule of thumb, unless the directions or your instructor says otherwise.
- Then, put a decimal point after the first digit!
- Count the number spaces you have after the decimal point.
- Now we get rid of all those zeros and add the notation “x 3”.
- The exponent on the 3, will simply be the number of spaces we had after the decimal point, which in our example was 12!
This works for small numbers too!
We just count the number of zeros that come AFTER the decimal point, and use a negative sign for our exponent.
Practice Problem!
Let’s say we have a Bacillus cell culture with a concentration of 6.2 x 104 cells/ml.
If I have 3 ml of the Bacillus cell culture and add 5 ml of TSB (nutrient broth) to the cell culture, what is my final concentration?
Practice Problem!
Practice Problem!
Practice Problem!
Practice Problem!
Practice Problem!
Practice Problem!
Practice Problem!
Practice Problem #2
What would our concentration be if we took an 8ml cell culture of bacillus and we added 3 ml of TSB broth? The cell culture has a concentration of 6.2 x 104 cells/ml.
Practice Problem #2
Once we have the number of cells, we get the final concentration, by dividing the number of cells by the final volume.
Practice Problem!
Practice Problem!
How do we count cells in culture?
The cells are way to small to just simply count directly!
If the culture is dilute enough, we could just mix our entire sample with warm agar...
And Then Incubate Overnight at Optimal Temperature, to Get Visible Colonies
We know one cell will yield one colony.
How Many Cells Were in the Original Sample?
How Many Cells Were in the Original Sample?
How Many Cells Were in the Original Sample?
Calculate the Concentration of Cells in Original Sample.
And Then Incubate Overnight at Optimal Temperature, to Get Visible Colonies
We know one cell will yield one colony.
BUT…. Here’s the problem! There can be millions of cells in a single drop! So, doing this is the without a serial dilution, will give you something more like…..
All of our previous ideas will work, once we get a sample of our cell culture that is dilute enough to yield between 30 and 300 colonies on a Petri plate/dish.
What if we did this same idea again, and just kept track of the volume?
We’ll Use Numbers that are Super Easy to Work With!
Setting up a serial dilution!
1:10
Start with 10 ml of your cell culture. This undiluted tube will be our tube #1. Then set up a number of tubes containing 9 ml each of broth alone (for dilutions).
Take 1 ml from the first tube and add it to the 9ml of plain broth you have in tube 2.
Take 1 ml from tube #2 and [place it into tube #3.
Tube #3 has 1 part that is from the 1:10 dilution, to its 10 ml total volume. So its ratio is 0.1/10 which reduces to 1/100. which = 0.01.
Take 1 ml from tube #3 and place it into tube #4.
Tube #4 has 1 ml from 1:100 which is 0.01 to its total volume of 10 ml, so its ratio is 0.01:10 = 𝟎.𝟎𝟏𝟏𝟎= 1:1000.
Take 1 ml from tube #4 and place it into tube #5.
Tube #5 has 1 ml from 1:1000 which is 0.001 to its total volume of 10 ml, so its ratio is 0.001:10 = 𝟎.𝟎𝟎𝟏𝟏𝟎= 1:10,000.
Take 1 ml from tube #5 and place it into tube #6.
Tube #6 has 1 ml from 1:10,000 which is 0.0001 to its total volume of 10 ml, so its ratio is 0.0001:10 = 𝟎.𝟎𝟎𝟎𝟏𝟏𝟎= 1:100,000.
Make a Pour Plate Cell Culture by Adding the Entire Contents of the Most Dilute Tube (Tube #6) by Adding it to Warm Agar in a Petri Dish.
Make sure to label your Petri dish with the dilution you used!
You should also make a culture of tube #5 and #4, just to be safe!
Incubate Overnight at Optimal Temperature
One progenitor cell or parent cell or culture-forming unit (CFU) will yield one single colony.
Now, see which of your culture plate contains between 30 – 300 colonies.
Ideally, you should have between 30 and 300 colonies in your dish.
Ideally, you should have one of your plates contain between 30 and 300 colonies.
COUNT THE COLONIES
# COLONIES = Count the bacterial colonies on the plate that grew between 30 and 300 colonies.
THE RECIPROCAL OF THE DILUTION FACTOR à
The Dilution Factor used for the plate with the colonies we counted,
= 𝟏𝟏𝟎,𝟎𝟎𝟎
To get the reciprocal, simply flip the fraction upside-down
= 𝟏𝟎,𝟎𝟎𝟎𝟏 = 10,000
Calculate the Number of Cells in Original Sample.
Calculate the Number of Cells in Original Sample.
Practice Problem #1
What is the concentration of a cell culture that grew 79 colonies on a Petri dish at a dilution of 1:100?
Practice Problem #1
What is the concentration of a cell culture that grew 79 colonies on a Petri dish at a dilution of 1:100?
Practice Problem #2
How many cell did we start with in our original cell culture?
Practice Problem #2
What is the concentration of a cell culture that grew 79 colonies on a Petri dish at a dilution of 1:100?
Calculate the Number of Cells in Original Sample.
QUESTION: Prepare a serial dilution to 1,000,000. SHORT CUT à There are 6 zeros in 1,000,000, so you’ll dilute it 6 times!
MATH CHEAT SHEET!
100 = 1 101 = 10 102 = 100 103 = 1000 104 = 10,000 105 = 100,000
106 = 1,000,000 107 = 10,000,000 108 = 100,000,000 109 = 1,000,000,000 1010= 10,000,000,000
100
MATH CHEAT SHEET!
10-1 = 0.1 = 1/10
10-2 = 0.01 = 1/100
10-3 = 0.001 = 1/1000
10-4 = 0.0001 = 1/10000
10-5 = 0.00001 = 1/100000
10-6 = 0.000001 = 1/1000000
10-7 = 0.0000001 = 1/10000000
10-8 = 0.00000001 = 1/100000000
10-9 = 0.000000001 = 1/1000000000
HOW TO SET UP A 3-FOLD DILUTION!
Setting up a serial dilution!
1:3
Start with 3 ml of your cell culture. This undiluted tube will be our tube #1. Then set up a number of tubes containing 2 ml each of broth alone (for dilutions).
Take 1 ml from the first tube and add it to the 2ml of plain broth you have in tube 2.
Take 1 ml from tube #2 and [place it into tube #3.
Tube #3 has 1 part that is from the 1:3 dilution, to its 3 ml total volume. So its ratio is 1/(3x3) = 1/9 or 1:9.
Take 1 ml from tube #3 and place it into tube #4.
Tube #4 has 1 ml from 1:9 to its total volume of 3 ml, so its ratio is = 𝟏𝟗 𝒙 𝟑= 1:27
Take 1 ml from tube #4 and place it into tube #5.
Tube #5 has 1 ml from 1:27 to its total volume of 3 ml, so its ratio is = 𝟏𝟐𝟕 𝒙 𝟑= 1:81
Take 1 ml from tube #5 and place it into tube #6.
Tube #6 has 1 ml from 1:81 to its total volume of 3 ml, so its ratio is = 𝟏𝟖𝟏 𝒙 𝟑= 1:243
Make a Pour Plate Cell Culture by Adding the Entire Contents of the Most Dilute Tube (Tube #6) by Adding it to Warm Agar in a Petri Dish.
Make sure to label your Petri dish with the dilution you used!
You should also make a culture of tube #5 and #4, just to be safe!
Incubate Overnight at Optimal Temperature
One progenitor cell or parent cell or culture-forming unit (CFU) will yield one single colony.
Now, see which of your culture plate contains between 30 – 300 cells.
Ideally, you should have between 30 and 300 colonies in your dish.
Ideally, you should have one of your plates contain between 30 and 300 colonies.
COUNT THE COLONIES
# COLONIES = Count the bacterial colonies on the plate that grew between 30 and 300 colonies.
THE RECIPROCAL OF THE DILUTION FACTOR à
The Dilution Factor used for the plate with the colonies we counted,
To get the reciprocal, simply flip the fraction upside-down
Calculate the Number of Cells in Original Sample.
1:10 Dilution Series
1:5 Dilution Series
1:2 Dilution Series
Ex. How many times would you need to do a two-fold serial dilution to get to 1:2048?
There’s an even faster way, but you’ll need a calculator.
Ex. How many times would you need to do a two-fold serial dilution to get to 1:2048?
Now go
CELL-ebrate your new skills!
THANK YOU FOR WATCHING!
Created by ScientistCindy.com
|
|
Escherichia coli is
|